Abstract
Purpose: In this study, the optimization of antitumor therapy with tumor necrosis factor-α (TNF-α) was attempted.
Experimental Design: Using the phage display technique, we created a lysine-deficient mutant TNF-α (mTNF-K90R). This mutant had higher affinities to both TNF receptors, despite reports that certain lysine residues play important roles in trimer formation and receptor binding.
Results: The mTNF-K90R showed an in vivo therapeutic window that was 13-fold higher than that of the wild-type TNF-α (wTNF-α). This was due to the synergistic effect of its 6-fold stronger in vitro bioactivity and its 2-fold longer plasma half-life derived from its surface negative potential. The reason why the mTNF-K90R showed a higher bioactivity was understood by a molecular modeling analysis of the complex between the wTNF-α and TNF receptor-I. The mTNF-K90R, which was site-specifically mono-PEGylated at the NH2 terminus (sp-PEG-mTNF-K90R), had a higher in vitro bioactivity and considerably longer plasma half-life than the wTNF-α, whereas the randomly mono-PEGylated wTNF-α had 6% of the bioactivity of the wTNF-α. With regard to effectiveness and safety, the in vivo antitumor therapeutic window of the sp-PEG-mTNF-K90R was 60-fold wider than that of the wTNF-α.
Conclusions: These results indicated that this functionalized TNF-α may be useful not only as an antitumor agent but also as a selective enhancer of vascular permeability in tumors for improving antitumor chemotherapy.
INTRODUCTION
With the success of the human genome project, the focus of life science research has shifted to the functional and structural analyses of proteins, such as the fields of proteomics and structural genomics. These analyses of proteins, including newly identified proteins, are expected to contribute to the identification of therapeutically applicable proteins for various diseases. Thus, pharmacoproteomic-based drug discovery and the development of protein therapies has currently attracted a great deal of attention (1, 2, 3). However, it is clinically difficult to use most bioactive proteins, such as tumor necrosis factor-α (TNF-α), as antitumor agents because of their very low stability and pleiotropic action in vivo (4, 5).
TNF-α was reported to exert a strong cytotoxicity to various kinds of tumor cells but not to normal cells in vitro and to cause hemorrhagic necrosis of certain transplanted solid tumors (6). Thus, TNF-α has been considered a promising new drug for cancer therapy. On account of its short plasma half-life, a continuous infusion or frequent administration at high doses of TNF-α was required to sustain its plasma level to obtain significant antitumor effects. Additionally, TNF-α was found to have unexpected toxic side-effects in phase I studies (7). These severe toxicities of TNF-α prevented the administration of dosages required for replicating the antitumor activity observed in preclinical studies. For this reason, the clinical application of TNF-α as a systemic antitumor agent has been limited, although intratumoral administration of TNF-α showed marked antitumor effects in phase I studies (8, 9). Recently, TNF-α has been clinically applied to locoregional combination therapy with Melphalan, and this therapy showed a marked antitumor effect in patients with in-transit melanoma metastases (10, 11, 12, 13). This clinical approach using TNF-α as a selective destruction agent against tumor endothelial cells and a selective enhancer of tumor vascular permeability for effective accumulation of antitumor chemotherapeutic agents is currently an attractive topic for optimization of cancer chemotherapy. This was approved by the European Agency for the Evaluation of Medicinal Products (14, 15). Additionally, fusion proteins composed of TNF-α and antitumor antibodies have been designed as systemic antitumor agents with tumor-targeting capabilities for improving TNF-α therapy (16, 17, 18, 19). Thus, the creation of a functionalized TNF-α having both superior antitumor effectiveness and safety will be useful for obtaining not only the required antitumor effects but also selectively enhancing the vascular permeability of tumor vessels by its systemic administration.
To additionally promote these clinical applications of TNF-α, it is necessary to develop a system to create mutant proteins (muteins) with desired properties, such as superior bioactivity, and a drug delivery system to selectively enhance the desirable therapeutic activities of TNF-α and its muteins without increasing their side effects. A protein-drug innovation system that fuses these two systems will demonstrate the tremendous potential of TNF-α therapy.
We have attempted previously to create useful muteins by the substitution of amino acids using a site-directed mutagenesis method, as typified by Kunkel’s method (20, 21). For instance, it was observed that a point mutation of basic residues that resulted in neutral or acid residues in immunotoxins lowered their isoelectric point, thus resulting in their reduced in vivo toxicity without loss of antitumor therapeutic activity. This was probably due to desirable changes in their pharmacokinetic properties that were derived from the surface negative electrostatic potential. However, the creation of muteins by such point mutation approaches takes a vast amount of time and effort to obtain the desired muteins, whereas the lowering of the isoelectric point of bioactive proteins may be an attractive approach to improve their therapeutic potency.
One of the most useful ways of enhancing the plasma half-lives of proteins is to conjugate them with polyethylene glycol (PEG) and other water-soluble polymeric modifiers (22, 23, 24, 25, 26, 27, 28). The covalent conjugation of proteins with PEG (PEGylation) increases their molecular size and steric hindrance, both of which depend on the properties of the PEG attached to the protein. This results in the avoidance of their renal excretion and in improvement of their proteolytic stability, whereas decreasing their immunogenicity and hepatic uptake. We have also reported that the optimal PEGylation of bioactive proteins could selectively improve their in vivo therapeutic potency and reduce side effects (22, 24, 25, 26). However, the PEGylation of proteins was mostly nonspecific and targeted at all of the lysine residues in the protein, some of which may be in or near an active site. As a result, the PEGylation of proteins was accompanied by a significant loss of their specific activities in vitro (27, 28). Thus, the clinical application of PEGylated proteins has been limited until today.
To overcome the problems of PEGylation mentioned above, using TNF-α as a model, we attempted recently to develop a novel strategy for site-specific mono-PEGylation for improvement of its in vivo antitumor potency (29). We isolated a bioactive lysine-deficient mutant TNF-α (mTNF-α) from phage libraries expressing mTNF-αs, in which all of the lysine residues were replaced with other amino acids. This lysine-deficient mTNF-α was site-specifically mono-PEGylated at its NH2 terminus with PEG. This site-specifically mono-PEGylated mTNF-α showed increased antitumor therapeutic potency, compared with the unmodified wild-type TNF-α (wTNF-α) and randomly mono-PEGylated wTNF-α. However, the in vitro bioactivity of this site-specifically mono-PEGylated mTNF-α and its affinity to both the TNF-receptor I (TNF-RI) and TNF-receptor II (TNF-RII) decreased significantly compared with those of the wTNF-α. Thus, if it was possible to create a lysine-deficient mTNF-α with stronger in vitro bioactivity and higher receptor-affinity, a site-specific PEGylation of such a super mutant TNF-α (smTNF-α), which has a lower isoelectric point than the wTNF-α, may synergistically enhance the antitumor activity in vivo without adverse side effects.
In this study, we attempted to create a lysine-deficient smTNF-α with superior in vitro bioactivity and a lower isoelectric point using the phage display technique with a few modifications. To our knowledge, there are no reports regarding the creation of lysine-deficient mutant proteins with a higher bioactivity. The molecular modeling of TNF-α revealed the mechanism of higher bioactivity of the obtained smTNF-α. The obtained smTNF-α was site-specifically PEGylated at its NH2 terminus. The mono-PEGylated smTNF-α thus obtained showed a higher in vitro bioactivity and longer plasma half-life than the unmodified wTNF-α and a previously reported site-specifically mono-PEGylated mTNF-α. Furthermore, its antitumor therapeutic window was 60-fold wider compared with that of the wTNF-α. Thus, this study will open new avenues for antitumor therapy using TNF-α as an antitumor agent and an enhancer of tumor vascular permeability. Additionally, this study may allow for the development of a novel protein-drug innovation system to promote pharmacoproteomic based drug discovery and development of protein therapies.
MATERIALS AND METHODS
Library Construction of Lysine-Deficient mTNF-αs and Selection of smTNF-α with Higher Bioactivity.
Plasmid pY02-TNF encoding the wTNF-α, in which the COOH terminus of TNF-α was fused to the NH2 terminus of the M13 phage g3p, was used as a PCR template for constructing the cDNA library of lysine-deficient mTNF-αs. A three-step PCR amplification was carried out using oligonucleotides containing the sequence “NNS” (where N and S represent G/A/T/C and G/C, respectively) at the wTNF-α codons for Lys11, Lys65, Lys90, Lys98, Lys112, and Lys128. The NNS can encode all 20 of the amino acids. The third PCR products were digested with restriction enzymes and ligated with the phagemid vector pY02. A phage library displaying lysine-deficient mTNF-αs was prepared, and phage clones displaying the smTNF-αs with a higher bioactivity were selected using BIAcore 2000 (BIAcore, Uppsala, Sweden) with TNF-RI, as described elsewhere (29). The bioactivity of each smTNF-α displayed by the phage clone was examined by a cytotoxicity assay using mouse LM cells, a cell line derived from L929 cells (30).
Expression and Purification of Recombinant TNF-αs.
Plasmids pYas1-TNF, pYas1-K90R, and pYas1-K90P encoding the human wTNF-α and smTNF-αs (mTNF-K90R and mTNF-K90P), under the control of a T7 promoter, were prepared. Three recombinant TNF-αs were produced in Escherichia coli BL21(DE3) harboring these expression plasmids as described previously (24, 29). Endotoxin levels were determined to be <300 pg/mg each in the wTNF-α and smTNFαs. The electrostatic potential surface was generated using GRASP (31). The TNF-α crystal structure, PDB entry 1TNF, was used for the calculation. The electrostatic potential ranged from −7.5 kT (red) to 7.5 kT (blue). The isoelectric point of each TNF-α was calculated using a program in the Genetics Computer Group (Madison, WI) package that is available online.7
PEGylation of TNF-αs.
Methoxy-PEG-succinimidyl propionate (mPEG-SPA; Mr 5000) was purchased from Shearwater Polymers (Huntsville, AL). The wTNF-α and mTNF-K90R in PBS were reacted with a 5-fold (wTNF-α) or 50-fold (mTNF-K90R) molar excess of mPEG5K-SPA, in terms of total primary amino groups of TNF-α, at 37°C for 30 minutes. After this, ε-aminocaproic acid (10 times molar excess in terms of the mPEG5K-SPA) was added to stop the reaction. The specific bioactivities of the mono-PEGylated forms of TNF-α were examined by a cytotoxicity assay using LM cells (30).
In vivo Studies.
All of the animal experiment protocols were in accordance with the Guide for Laboratory Animal Facilities and Care. These protocols have been approved by the committee of the Pharmaceutical School, Osaka University. The antitumor effects of TNF-αs were assessed in mice bearing Meth-A fibrosarcoma. The Meth-A cells were implanted intradermally (2 × 105 cells per site) in 5-week-old female BALB/c mice. On day 7, when the tumor diameter reached 7 mm, the TNF-α molecules were administered by a single i.v. injection. The antitumor potency was estimated from the tumor volume and tumor hemorrhagic necrosis 24 hours after the injection. The tumor volume was calculated using the formula described by Haranaka et al. (32). For the pharmacokinetic assay, normal female BALB/c mice were injected i.v. with 1 μg each of various TNF-αs. Blood samples were drawn at different times after the injection. The concentrations of TNF-α in the blood samples were measured by ELISA. For assessment of in vivo toxicity (LD50; the dose that kills half of the animals tested), groups of 4 to 6 female BALB/c mice were injected i.v. with increasing doses of various TNF-αs.
RESULTS
Selection of a Lysine-Deficient smTNF-α with Higher Bioactivity.
To create a lysine-deficient smTNF-α with higher bioactivity, a phage library displaying mTNF-αs with randomized sequences in place of the six lysine codons was prepared. The phage library was subjected to several rounds of panning against human TNF-RI using a BIAcore biosensor, and the clones were screened for TNF-specific bioactivity by a cytotoxicity assay using LM cells. In this screening, we succeeded in obtaining two smTNF-αs (mTNF-K90R and mTNF-K90P). DNA sequencing analysis of the mTNF-K90R and mTNF-K90P indicated that these proteins lacked all six of the lysine residues, which replaced similar amino acids (Table 1). The isoelectric point of the mTNF-K90R and mTNF-K90P was lowered from 7.44 (wTNF-α) to 4.96 and 4.76, respectively.
Properties of the Novel Lysine-Deficient smTNF-αs.
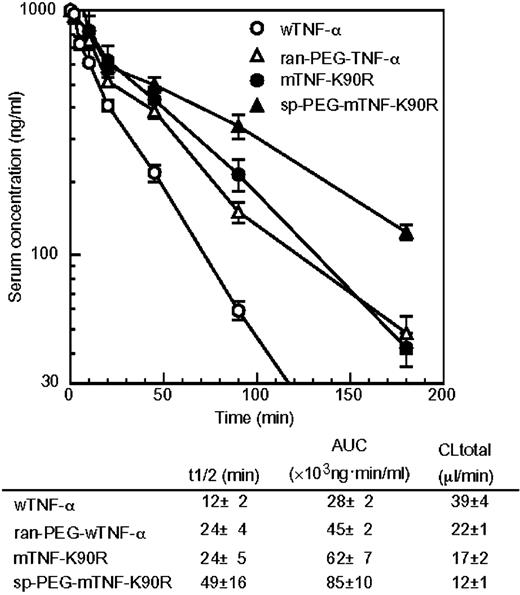
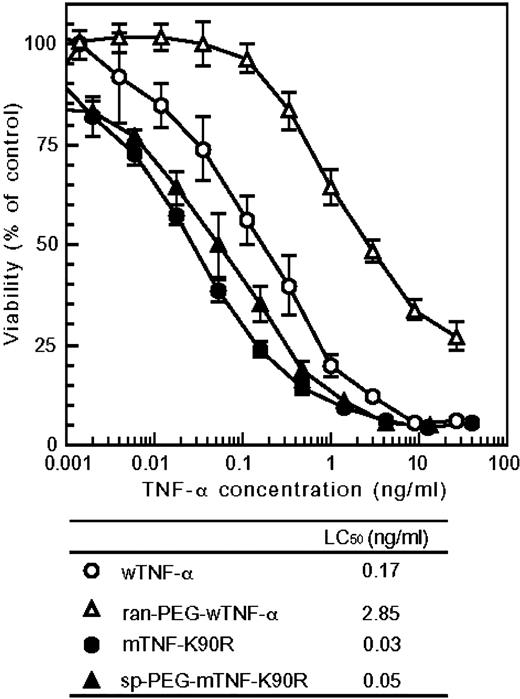
The recombinant human wTNF-α, mTNF-K90R, and mTNF-K90P were prepared by a general recombinant protein technology. The purified mTNF-K90R and mTNF-K90P were analyzed by SDS-PAGE that revealed a single band of ∼17 KDa as well as the wTNF-α. Using gel filtration chromatography, we confirmed that the mTNF-K90R and mTNF-K90P formed a trimeric structure, similar to that of the wTNF-α and natural human TNF-α, in an aqueous solution (data not shown). We assessed the changes in the surface electrostatic potential of both the mTNF-K90R and mTNF-K90P. Fig. 1 shows the electrostatic potential mapped onto the molecular surface of the wTNF-α, mTNF-K90R, and mTNF-K90P with red designating negative and blue designating positive potential values. The surface areas of negative potential were found to increase on the mTNF-K90R and mTNF-K90P due to the lack of lysine residues. The Kd value of the mTNF-K90R and mTNF-K90P to TNF-RI was 1.3-fold stronger than that of the wTNF-α, as shown in Table 1. The affinity of this smTNF-α to TNF-RII was also higher than that of the wTNF-α (data not shown). By means of the in vitro cytotoxicity assay using mouse LM cells in the presence of actinomycin D at the concentration of 2 μg/mL, the LC50 values (specific bioactivity) of the wTNF-α, mTNF-K90R, and mTNF-K90P were found to be 0.17 ng/mL, 0.03 ng/mL, and 0.14 ng/mL, respectively. We constructed >10 mutants of the mTNF-K90R in which K90 replaced any other residues by site-directed mutagenesis. The mTNF-K90R had the highest in vitro bioactivity and affinity to both TNF-RI and TNF-RII. The in vivo antitumor activity of the mTNF-K90R against mouse Meth-A solid tumors after a single i.v. injection was assessed by evaluating tumor hemorrhagic necrosis as an index (Fig. 2,A). The wTNF-α showed weak hemorrhagic necrotic effects in a dose-dependent manner. In contrast, the mTNF-K90R at a dose of 0.3 μg per mouse expressed antitumor effects superior to those of the wTNF-α at a dose of 3 μg per mouse. The LD50 values for the wTNF-α and mTNF-K90R were 390 and 510 μg protein/kg, respectively (Table 1). These results indicated that the mTNF-K90R had 10-fold stronger in vivo antitumor activity and 1.3-fold weaker toxicity than the wTNF-α, whereas the in vitro bioactivity of the mTNF-K90R was 6-fold stronger. Thus, the pharmacokinetics of the mTNF-K90R after the i.v. injection of a single dose of 1 μg per mouse were measured (Fig. 3). We found that the plasma half-life of the mTNF-K90R was 2-fold longer than that of the wTNF-α.
Site-specific PEGylation of mTNF-K90R.
The wTNF-α and mTNF-K90R were modified with 5 kDa of activated PEG. We confirmed by SDS-PAGE that a single PEG molecule was attached selectively to the NH2 terminus of the mTNF-K90R, whereas PEG molecules were introduced randomly at multiple positions in the wTNF-α (data not shown). TNF-α molecules conjugated with one PEG molecule were purified by gel filtration- high-performance liquid chromatography. The purified site-specific mono-PEGylated mTNF-K90R (sp-PEG-mTNF-K90R) and randomly mono-PEGylated wTNF-α (ran-PEG-wTNF-α) were examined for their specific bioactivity (Fig. 4). The sp-PEG-mTNF-K90R had 60% of the specific activity of the mTNF-K90R, whereas the ran-PEG-wTNF-α had only 6% of the specific activity of the wTNF-α. Surprisingly, the sp-PEG-mTNF-K90R had higher in vitro bioactivity than the wTNF-α.
Antitumor Therapeutic Window of the sp-PEG-mTNF-K90R.
To clarify the antitumor therapeutic window of the sp-PEG-mTNF-K90R, its in vivo antitumor activity and toxicity were assessed (Fig. 2). As shown in Fig. 2,A, the antitumor activity of the ran-PEG-wTNF-α was similar to that of the wTNF-α. In contrast, the sp-PEG-mTNF-K90R at a dose of 1 μg per mouse induced marked tumor hemorrhagic necrosis compared with the mTNF-K90R at a dose of 3 μg per mouse. These results indicated that the antitumor activity of the sp-PEG-mTNF-K90R was >3-fold higher than that of the mTNF-K90R, which had in vivo antitumor effects that were 10-fold stronger than the wTNF-α. Thus, the sp-PEG-mTNF-K90R had an antitumor potency that was >30-fold higher than that of the wTNF-α and ran-PEG-wTNF-α. A single i.v. injection of the sp-PEG-mTNF-K90R at a dose of 1 μg per mouse completely inhibited solid tumor growth (Fig. 2,B). The LD50 values of the sp-PEG-mTNF-K90R and ran-PEG-wTNF-α were 780 and 1,290 μg protein/kg, respectively. These results indicated that the in vivo toxicity of the sp-PEG-mTNF-K90R was ∼1.5-fold, 2.0-fold, and 0.6-fold lower than that of the mTNF-K90R, wTNF-α, and ran-PEG-wTNF-α, respectively. Thus, the therapeutic window of the sp-PEG-mTNF-K90R expanded by >5-fold, 60-fold, and 18-fold compared with that of the mTNF-K90R, wTNF-α, and ran-PEG-wTNF-α, respectively. The pharmacokinetics of the sp-PEG-mTNF-K90R after i.v. injection were also assessed (Fig. 3). The plasma half-life of the sp-PEG-mTNF-K90R was 49 minutes, which was longer than that of the mTNF-K90R (24 minutes), wTNF-α (12 minutes), and ran-PEG-wTNF-α (24 minutes).
DISCUSSION
Several therapeutically useful bioactive proteins have been identified in the postgenome era. However, as elucidated in the clinical trials of various proteins in the past, protein therapy still has many problems derived from the in vivo drawbacks of proteins, such as their in vivo low stability (short plasma half-life) and complicated actions. One way to circumvent this problem may be to synthesize functional muteins by traditional techniques of amino acid substitution, such as site-directed mutagenesis, based on the structural simulation data of a mutein (21, 33, 34). However, it is difficult to obtain muteins with desired properties using these methods. This is because useful muteins must be identified only among several kinds of structural variant proteins that are produced by trial and error, after a great deal of time. Another effective method to overcome the in vivo drawbacks of proteins may be to modify the proteins with PEG (35, 36). However, the application of PEGylation has been limited to a small part of the protein, because the random introduction of PEG to ε-amino groups usually lowers the bioactivity of proteins markedly. Thus, the improvement of both the protein-drug innovation systems mentioned above is indispensable for the promotion of protein therapy. In this study, using TNF-α, we attempted to develop a novel protein-drug innovation system by fusing technology used to create clinically useful muteins and site-specific PEGylation.
Phage libraries displaying polypeptides, such as naive antibodies or random peptides, have extensively been applied for the identification of specific molecules with a high affinity for a target ligand (37, 38, 39). The advantages of a phage display system are easy preparation of a library consisting of structural variants of a polypeptide as diverse as over one hundred million and isolation of several molecules binding to a targeted ligand from this library in a few weeks. However, there are few studies on the application of the phage display technique for creation of therapeutically useful structural variants of a bioactive protein, such as muteins with a stronger bioactivity and longer plasma half-life. To create a lysine-deficient smTNF-α with a stronger in vitro bioactivity than the wTNF-α, a phage library displaying a lysine-deficient mTNF-α was prepared, and it consisted of ∼1 × 108 independent structural variants. After two rounds of biopanning against TNF-RI, the mTNF-K90R with an in vitro bioactivity that was 6-fold stronger was obtained, despite reports that some lysine residues were essential for its bioactivity (Fig. 2 and Table 1; refs. 40, 41, 42, 43). This mTNF-K90R has an ∼10-fold higher in vivo antitumor potency and 1.3-fold lower in vivo toxicity compared with that of wTNF-α. Therefore, the therapeutic window of the mTNF-K90R was extended by ∼13-fold compared with that of the wTNF-α (Fig. 2,A and Table 1). This improved therapeutic window of the mTNF-K90R was due to its stronger in vitro bioactivity as well as its longer plasma half-life (Fig. 3). We reported previously that lowering the isoelectric point of antitumor immunotoxins increased their therapeutic potency, probably due to the desirable changes in their pharmacokinetic properties derived from the surface negative electrostatic potential (21). Thus, the longer half-life of the mTNF-K90R may partially result from the effects of lowering the isoelectric point.
Our data are the first to report the creation of a lysine-deficient mutant protein with a higher bioactivity and enhanced therapeutic antitumor effects. It is interesting to examine, from the standpoint of structure, why the mTNF-K90R had a stronger in vitro bioactivity and higher affinity to TNF-RI than the wTNF-α, when all of the lysine residues in the mTNF-K90R are replaced with other amino acids. Site-directed mutagenesis analysis of TNF-α suggested that among six lysine residues, Lys65 and Lys90, in particular, were involved in the interaction with its receptor (40, 41, 42, 43). It was shown that the TNF-α mutant, in which Lys65 was replaced by Ala65 (K65A), bound the receptor better than the wTNF-α, whereas the K65W mutant showed a remarkably reduced binding to the receptor. In the wTNF-α structure, it was predicted by molecular modeling of the complexes between TNF-α and TNF-RI that Lys65 would repel against Lys78 in TNF-RI (Fig. 5,B). This predicted model explains that the increased binding affinity of K65A would be due to the absence of short contacts between Ala65 in K65A and Lys78 in TNF-RI, and the decreased affinity of K65W may result from steric interference between Trp65 in K65W and Lys78 in TNF-RI. In the mTNF-K90R, Lys65 was replaced by Ser65. We considered that the substitutions of Lys65 with a small amino acid, such as Ser65 in the mTNF-K90R, would enable these proteins to bind receptors because of the loss of interference between Lys65 in TNF-α and Lys78 in TNF-RI. Surprisingly, Lymphotoxin-α (TNF-β), which can bind TNF-RI with similar affinity to TNF-α, has Ser65 instead of Lys65 in TNF-α (44). In the wTNF-α structure, Lys90 forms a hydrogen bond with Glu135 (Fig. 5 C). This interaction is likely to stabilize the loop structure containing residues 84 to 89, which are involved in the receptor binding in the model. The loop is located near the trimer interface, and receptor binding studies have also suggested that the loop is essential for receptor binding. In the mTNF-K90R, Arg90 is also likely to be involved in hydrogen bonding with Glu135. The interaction would contribute to the stabilization of the loop structure. To additionally investigate the relationship between structure and activity, we are attempting to reveal the crystal structure of the mTNF-K90R.
The PEGylation of proteins is mostly nonspecific and may occur at all of the lysine residues, some of which may be within or near an active site. The resultant PEGylated proteins show markedly lower bioactivity. Additionally, the PEGylated proteins are composed of positional isomers with PEG at various sites, which have distinct activities and other characteristics (45). The NH2 terminus site-specific mono-PEGylation was found to improve the plasma half-life of the mTNF-K90R without a marked reduction of its in vitro bioactivity, whereas the ran-PEG-wTNF-α had 6% of the bioactivity of the wTNF-α. Additionally, the sp-PEG-mTNF-K90R with superior molecular uniformity showed a much higher in vitro bioactivity than the wTNF-α and a site-specific PEGylated mutant TNF-α reported previously. Our data are the first to report the creation of a PEGylated protein with a higher bioactivity than the unmodified parent protein. Thus, these results indicate that this site-specific PEGylation system has solved the problems of previous random PEGylation systems. The sp-PEG-mTNF-K90R had a 3-fold higher in vivo antitumor effect and 1.5-fold lower toxicity compared with the mTNF-K90R; thus, the therapeutic window of the sp-PEG-mTNF-K90R was enhanced by 4.5-fold and 60-fold compared with that of the mTNF-K90R and wTNF-α, respectively. This may be due to the longer plasma half-life of the sp-PEG-mTNF-K90R. However, the replacement of the lysine residues in the mTNF-K90R may increase the immunogenicity of the protein, which must be investigated, although on the other hand, PEGylation is known to considerably decrease protein immunogenicity (24). When TNF-α is used as a systemic antitumor agent, its dose must be restricted to only 1/5 to 1/25 of the amount necessary to obtain sufficient antitumor activity, due to its adverse side-effects. The therapeutic window of the sp-PEG-mTNF-K90R was found to be 60-fold wider than that of the wTNF-α. TNF-α in combination with Melphalan achieved improved tumor response using local perfusion in transit melanoma metastases and limb salvage in soft-tissue sarcoma patients. This is because TNF-α selectively injured tumor endothelial cells and enhanced the vascular permeability of tumor vessels (10, 11, 12, 13, 14, 15). The sp-PEG-mTNF-K90R can be used not only for systemic administration but also in combination with chemotherapy. In addition, some researchers reported that fusion of TNF-α with a targeting ligand such as an antibody and peptide, against tumor or tumor endothelial cells, was useful for therapy (16, 17, 18, 19). Currently, we are attempting to apply this system for creating muteins and for site-specific PEGylation of the fusion TNF-α.
In this study, we showed the advantage of a mutein creation system using the phage display technique and a site-specific PEGylation system to produce lysine-deficient muteins with a superior bioactivity for the promotion of pharmacoproteomic-based protein-drug discovery and development. The fusion of these two systems may be, at present, the best way to design optimal protein-drugs for therapy.
Grant support: Grant-in-Aid for Scientific Research (No. 15680014 and No. 16023242) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, in part by a Health and Labor Sciences Research Grants from Ministry of Health, Labor, and Welfare of Japan, in part by Health Sciences Research Grants for Research on Health Sciences Focusing on Drug Innovation from the Japan Health Sciences Foundation, in part by Takeda Science Foundation, and in part by Senri Life Science Foundation.
Note: H. Shibata and Y. Yoshioka contributed equally to the work.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Requests for reprints: Yasuo Tsutsumi, Department of Biopharma-ceutics, Graduate School of Pharmaceutical Sciences, Osaka Uni-versity, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan. Phone and Fax: 81-6-6879-8178; E-mail: ytsutsumi@nihs.go.jp or tsutsumi@phs.osaka-u.ac.jp
Internet address: http://molbio.info.nih.gov/molbio/gcglite/protform.htm.