


Abstract
Purpose: Overexpression of the multidrug resistance proteins P-glycoprotein (Pgp), multidrug resistance protein (MRP-1), breast cancer resistance protein (BCRP), and lung resistance protein (LRP) is associated with treatment failure in acute myeloid leukemia (AML) and other malignancies. The Pgp modulator cyclosporin A has shown clinical efficacy in AML, whereas its analogue PSC-833 has not. Cyclosporin A is known to also modulate MRP-1, and we hypothesized that broad-spectrum multidrug resistance modulation might contribute to its clinical efficacy.
Experimental Design: We studied the effects of cyclosporin A and PSC-833 on in vitro drug retention and cytotoxicity in resistant cell lines overexpressing Pgp, MRP-1, and BCRP and on nuclear-cytoplasmic drug distribution and cytotoxicity in cells overexpressing LRP. Cellular drug content was assessed by flow cytometry and nuclear-cytoplasmic drug distribution by confocal microscopy.
Results: Cyclosporin A enhanced retention of the substrate drug mitoxantrone in cells overexpressing Pgp (HL60/VCR), MRP-1 (HL60/ADR), and BCRP (8226/MR20, HEK-293 482R) and increased cytotoxicity 6-, 4-, 4-, and 3-fold, respectively. Moreover, cyclosporin A enhanced nuclear distribution of doxorubicin in 8226/MR20 cells, which also express LRP, and increased doxorubicin cytotoxicity 12-fold without an effect on cellular doxorubicin content, consistent with expression of wild-type BCRP, which does not efflux doxorubicin. Cyclosporin A also enhanced nuclear doxorubicin distribution in a second cell line with LRP overexpression, HT1080/DR4. PSC-833 enhanced mitoxantrone retention and cytotoxicity in cells overexpressing Pgp, but had no effect in cells overexpressing MRP-1, BCRP, or LRP.
Conclusions: Cyclosporin A modulates Pgp, MRP-1, BCRP, and LRP, and this broad-spectrum activity may contribute to its clinical efficacy.
INTRODUCTION
Overexpression of the multidrug resistance (MDR) protein P-glycoprotein (Pgp), an ATP-binding cassette (ABC) protein that mediates energy-dependent drug efflux, has been associated with treatment failure in diverse malignancies including acute myelogenous leukemia (AML; refs. 1–3). Consequently, administration of MDR modulators that block drug efflux mediated by Pgp in combination with chemotherapy is being explored as a novel treatment strategy.
Agents tested as clinical MDR modulators have included cyclosporin A and its nonimmunosuppressive, nonnephrotoxic analogue PSC-833, a potent Pgp modulator in preclinical models (4). A randomized clinical trial of cyclosporin A in refractory and relapsed AML showed significant prolongation of disease-free survival on the cyclosporin A arm (5). In contrast, similar benefit has not been seen with PSC-833 in randomized trials (6, 7), although in one of the trials a trend toward longer disease-free survival was seen on the PSC-833 arm in patients in whose AML cells PSC-833 modulated drug efflux in vitro (6).
Other MDR-associated ABC proteins that are expressed in AML and other malignancies and that also appear to impact treatment outcome include multidrug resistance protein (MRP-1) and breast cancer resistance protein (BCRP; refs. 8–11). All AML samples studied to date have expressed wild-type BCRP (BCRPR482; refs. 11, 12), which mediates mitoxantrone resistance but only low-level anthracycline resistance, whereas BCRP with mutations changing arginine at amino acid 482 to threonine (BCRPR482T) or glycine (BCRPR482G) mediates resistance to both mitoxantrone and anthracyclines (13). AML cells also express lung resistance protein (LRP; ref. 14), a vault protein that appears to block cytoplasmic-nuclear drug transport (15, 16).
Cyclosporin A is known to have activity as a modulator of MRP-1, in addition to Pgp (17, 18). We hypothesized that the clinical efficacy of cyclosporin A as a modulator in AML might in part reflect a broad spectrum of activity against the MDR proteins expressed in AML cells. We therefore compared the effects of cyclosporin A and PSC-833 on drug retention and cytotoxicity in resistant cell lines overexpressing Pgp, MRP-1, and BCRP and on cytoplasmic-nuclear drug distribution and cytotoxicity in cells expressing LRP.
MATERIALS AND METHODS
Cell Lines. Drug-selected cell lines overexpressing Pgp (HL60/VCR, A2780/Dx5b), MRP-1 (HL60/Adr), BCRPR482 (8226/MR20), BCRPR482T (MCF7 AdVp3000), or BCRPR482G (S1M180) were studied, along with parental HL60 cells, which express none of the proteins, as previously described (19, 20), and parental A2780, MCF7, and S1 cells. HEK-293 cells transfected with BCRPR482, BCRPR482T, BCRPR482G, or plasmid vector (482R, 482G, 482T, pcDNA), obtained from Dr. Susan E. Bates (National Cancer Institute, Bethesda, MD) were also studied (13). The drug-selected cell line 8226/MR20 also overexpresses LRP (14, 21) and therefore was also used in the experiments testing modulation of LRP, with parental 8226/S cells as a control. HT1080/DR4 fibrosarcoma cells, which express LRP and MRP-1, were studied as a second LRP-expressing cell line, with parental HT1080 cells as a control (22).
Drugs. Mitoxantrone (Sigma, St. Louis, MO), a substrate for Pgp, MRP-1, BCRPR482, BCRPR482T, and BCRPR482G, was used in the drug uptake experiments and cytotoxicity assays, as previously described (19, 20, 23). Doxorubicin (Sigma), a substrate for Pgp, MRP-1, BCRPR482T, and BCRPR482G, but not BCRPR482, was used in confocal microscopy experiments and was also used in the corresponding cytotoxicity assays.
Modulators. Cyclosporin A (Novartis Pharmaceutical Corporation, East Hanover, NJ) and PSC-833 (Novartis) were compared with the MRP-1–specific modulator MK571 (Merck Frosst Canada Ltd., Quebec, Canada; ref. 24) and the BCRP-specific modulator fumitremorgin C (23), a gift from Dr. Susan Bates. Cyclosporin A and PSC-833 were studied at a concentration of 2.5 μmol/L (6, 25), fumitremorgin C at 10 μmol/L (23), and MK571 at 5 μmol/L (24).
Drug Uptake Assay and Data Analysis. A total of (0.5 to 1) × 106 cells were incubated with 3 μmol/L mitoxantrone for 30 minutes at 37°C in the presence and absence of modulator, washed with cold PBS, and kept on ice until analysis. Mitoxantrone content was measured on a FACScan flow cytometer (Becton Dickinson, San Jose, CA) and analyzed using WinList software (Verity Software House, Topsham, ME), as previously described (23). Mitoxantrone content after uptake with and without modulator was compared using the Kolmogorov-Smirnov statistic, expressed as a D value ranging from 0 (no difference) to 1 (no overlap), as previously described (23). Based on previous work, D values ≥0.2 indicate material modulation (23). Statistically significant differences between D values for modulated cell lines expressing ABC transporters and an HL60 negative control (not expressing any ABC transporter), or between D values for rival modulators for a specific cell line, were determined with a two-sample t test (P < 0.05). No adjustments were made for multiple comparisons. All t tests were done with the Minitab software package (State College, PA).
Cytotoxicity Assays and Data Analysis. To study cytotoxicity, suspension cell lines were plated in 96-well tissue culture plates at a density of 10,000 cells per well in RPMI 1640 supplemented with 10% FCS, 2 mmol/L l-glutamine, 20 units/mL penicillin, and 20 μg/mL streptomycin, and drug was added to the culture medium to achieve final concentrations of 0.3 nmol/L to 10 μmol/L, with half-log increments, with and without modulator. The final volume of medium per well was 100 μL. Adherent cell lines were seeded at 600 to 2,000 cells per well (cell line dependent) in 96-well plates and incubated for 18 to 24 hours at 37°C to allow for attachment and then similarly treated. Cells were incubated for 96 hours at 37°C in a fully humidified atmosphere of 5% CO2 in air. Cell growth was assessed by the WST-1 colorimetric assay (Roche Diagnostics GmbH, Mannheim, Germany) done according to the manufacturer's instructions. Briefly, 10 μL of WST-1 were added to each well and culture plates were returned to the incubator for an additional 4 hours. Subsequently, the absorbance at 450 nm (A450) and 600 nm (A600, background) was read for each well using a Microtek multiwell plate reader. Each drug exposure condition was assessed in quadruplicate.
IC50s were determined by fitting the four-parameter Hill model to data with nonlinear regression, and resistance-modifying factors (RMF) were calculated as the ratios IC50 drug/IC50 drug + modulator, as previously described (19, 20). Ratios >1 indicate modulation. Significant differences in RMF from 1 were determined with a one-sample t test (P < 0.05) after a base 10 logarithmic transformation of the RMF values. Experiments were done in triplicate except for the MCF7 AdVp3000 experiments, which were done in duplicate, and results are reported as geometric mean values. Differences in RMFs between modulators were determined with a two-sample t test (P < 0.05) after a base 10 logarithmic transformation of the RMF values. No adjustments were made for multiple comparisons. Logarithmic transformations of RMF values are necessary before the calculation of some summary measures and the conduct of some statistical tests because logical differences in RMFs are multiplicative (not additive). Therefore, the log transform is required to change multiplicative differences into additive differences, making the data conform to the key linearity assumption of the t tests.
Nuclear Factor–κB Expression and Cellular Localization. Cellular expression of nuclear factor–κB (NF–κB) was studied by flow cytometry using a phycoerythrin-conjugated monoclonal antibody specific for the NF–κB p65 subunit (Santa Cruz Biotechnology, Santa Cruz, CA) in cells fixed in 3.7% paraformaldehyde, then 90% methanol for 10 minutes each. After flow cytometric analyses, the cells were also studied by confocal microscopy to determine cellular localization of NF–κB.
Intracellular Distribution of Doxorubicin. Confocal microscopy was used to study modulation of nuclear-cytoplasmic drug distribution in cells expressing LRP (26, 27). Confocal microscopy was done as previously described (20). Briefly, 1 × 106 cells were incubated for 3 hours with 5 μmol/L doxorubicin with and without modulator(s), washed, and aliquots placed onto Alcian blue–coated slides and studied by confocal microscopy. The excitation light source was set at 488 nm, and emission was captured through a 550-nm long-pass filter. For each cell, 10 to 20 focal planes were evaluated, and images with optimal nuclear-cytoplasmic ratios were stored for analysis.
RESULTS
To show that neither cyclosporin A nor PSC-833 is cytotoxic at the concentration used, cells from each cell line were cultured for 96 hours in 96-well plates in medium without modulator, and with 2.5 μmol/L cyclosporin A or 2.5 μmol/L PSC-833. For all cell lines, <10% difference was observed between growth in the presence and absence of cyclosporin A or PSC-833 (data not shown). In addition, neither PSC-833 nor cyclosporin A altered mitoxantrone retention (Fig. 1A) nor changed the IC50 of mitoxantrone in parental HL60 cells, which do not express any of the MDR-associated proteins, nor in parental A2780, MCF7, or S1 cells (Fig. 1B).

A, modulation of mitoxantrone uptake by cyclosporin A (CsA) and PSC-833 in resistant cell lines overexpressing Pgp, MRP-1, BCRPR482, BCRPR482T, and BCRPR482G. D values compare histograms measuring mitoxantrone content after uptake with and without modulator. Columns, means of triplicate (MCF7 AdVp3000 duplicate) experiments. HL60 cells are shown on the left as a negative control. ∩, P < 0.05 (significant difference between the two modulators for a specific cell line); *, P < 0.05 (significant difference between the modulated resistant cell line and the respective modulated HL60 control; ‡, P > 0.05 but < 0.1. B, modulation of mitoxantrone cytotoxicity by cyclosporin A and PSC-833 in resistant cell lines overexpressing Pgp, MRP-1, BCRPR482, BCRPR482T, and BCRPR482G and in parental cells. The RMF is calculated as the ratio IC50 drug/IC50 drug + modulator. Columns, geometric means of triplicate (MCF7 AdVp3000 duplicate) experiments. ∩, P < 0.05 (significant difference between the two modulators for a specific cell line); *, P < 0.05 (significant difference in RMF between a modulated cell line and RMF = 1; ‡, P > 0.05 but < 0.1.
A, modulation of mitoxantrone uptake by cyclosporin A (CsA) and PSC-833 in resistant cell lines overexpressing Pgp, MRP-1, BCRPR482, BCRPR482T, and BCRPR482G. D values compare histograms measuring mitoxantrone content after uptake with and without modulator. Columns, means of triplicate (MCF7 AdVp3000 duplicate) experiments. HL60 cells are shown on the left as a negative control. ∩, P < 0.05 (significant difference between the two modulators for a specific cell line); *, P < 0.05 (significant difference between the modulated resistant cell line and the respective modulated HL60 control; ‡, P > 0.05 but < 0.1. B, modulation of mitoxantrone cytotoxicity by cyclosporin A and PSC-833 in resistant cell lines overexpressing Pgp, MRP-1, BCRPR482, BCRPR482T, and BCRPR482G and in parental cells. The RMF is calculated as the ratio IC50 drug/IC50 drug + modulator. Columns, geometric means of triplicate (MCF7 AdVp3000 duplicate) experiments. ∩, P < 0.05 (significant difference between the two modulators for a specific cell line); *, P < 0.05 (significant difference in RMF between a modulated cell line and RMF = 1; ‡, P > 0.05 but < 0.1.
Both cyclosporin A and PSC-833 modulated mitoxantrone uptake (Fig. 1A) and cytotoxicity (Fig. 1B) in the cell lines that overexpressed Pgp (HL60/VCR and A2780/Dx5b). In addition, cyclosporin A also modulated mitoxantrone uptake (Fig. 1A) and cytotoxicity (Fig. 1B) in cell lines that expressed MRP-1 (HL60/ADR), BCRPR482 (8226/MR20 and HEK-293 482R), and BCRPR482T (MCF7-AdrVp3000 and HEK-293 482T). Finally, cyclosporin A modulated cytotoxicity in the cell lines that expressed BCRPR482G (S1M180 and HEK-293 482G), although an effect on uptake was only seen in HEK-293 482G cells. In contrast to cyclosporin A, PSC-833 had minimal or no effect on mitoxantrone uptake (Fig. 1A) and minimal or no effect on cytotoxicity (Fig. 1B) in cells that expressed MRP-1, BCRPR482, BCRPR482T, or BCRPR482G.
Modulation by cyclosporin A was compared with modulation by the ABC protein–specific modulators MK571 (MRP-1) and fumitremorgin C (BCRP) in resistant cell lines overexpressing MRP-1 and BCRP, respectively (Table 1). The RMFs seen with cyclosporin A were smaller than those seen with MK571 and fumitremorgin C in the cells that overexpressed MRP-1 and BCRPR482, the ABC proteins known to be expressed in AML and other malignancies. The RMFs of cyclosporin A were not different from the RMFs of fumitremorgin C in cells transfected with BCRPR482T and BCRPR482G, but were smaller than those of the drug-selected cell lines that overexpressed these proteins, which have not been found in AML or in other malignancies to date.
RMFs of cyclosporin A compared with those of the ABC protein–specific modulators MK571 and fumitremorgin C in resistant cell lines overexpressing MRP-1 and BCRP, respectively
| Protein | Cell line | CsA | MK571 | FTC |
|---|---|---|---|---|
| MRP-1 | HL60/Adr* | 4.07 (3.74 to 4.43) | 8.90 (6.88 to 11.5) | |
| BCRPR482 | HEK-293 482R† | 3.11 (0.789 to 12.2) | 9.44 (6.69 to 13.3) | |
| 8226/MR20† | 4.12 (1.49 to 11.4) | 9.89 (5.83 to 16.7) | ||
| BCRPR482T | HEK-293 482T | 5.20 (3.19 to 8.47) | 4.69 (3.12 to 7.06) | |
| MCF7 AdVp3000† | 5.68 (0.996 to 32.4) | 35.5 (8.26 to 151) | ||
| BCRPR482G | HEK-293 482G | 4.25 (1.46 to 12.3) | 5.20 (2.72 to 9.95) | |
| S1M180* | 4.01 (3.59 to 4.47) | 77.6 (46.8 to 129) |
| Protein | Cell line | CsA | MK571 | FTC |
|---|---|---|---|---|
| MRP-1 | HL60/Adr* | 4.07 (3.74 to 4.43) | 8.90 (6.88 to 11.5) | |
| BCRPR482 | HEK-293 482R† | 3.11 (0.789 to 12.2) | 9.44 (6.69 to 13.3) | |
| 8226/MR20† | 4.12 (1.49 to 11.4) | 9.89 (5.83 to 16.7) | ||
| BCRPR482T | HEK-293 482T | 5.20 (3.19 to 8.47) | 4.69 (3.12 to 7.06) | |
| MCF7 AdVp3000† | 5.68 (0.996 to 32.4) | 35.5 (8.26 to 151) | ||
| BCRPR482G | HEK-293 482G | 4.25 (1.46 to 12.3) | 5.20 (2.72 to 9.95) | |
| S1M180* | 4.01 (3.59 to 4.47) | 77.6 (46.8 to 129) |
NOTE. Results are shown as geometric means of triplicate experiments, with 95% confidence intervals around the geometric mean.
Abbreviation: CsA, cyclosporin A; FTC, fumitremorgin C.
P < 0.05, statistically significant difference between RMFs.
P > 0.05 but < 0.1.
To look for possible differential effects of cyclosporin A and PSC-833 on NF–κB expression and/or localization that could contribute to their differential effects on mitoxantrone cytotoxicity, expression and nuclear/cytoplasmic localization of NF–κB p65 were studied in HL60 cells. Neither cyclosporin A nor PSC-833 affected the expression level or cellular localization of NF–κB p65, nor did cyclosporin A alter the nuclear relocalization induced by mitoxantrone (Fig. 2).

Effect of cyclosporin A, PSC-833, and mitoxantrone on NF–κB expression levels and cellular localization. NF–κB expression levels, studied by flow cytometry, were not affected by exposure to cyclosporin A, PSC-833, mitoxantrone or the combination of mitoxantrone + cyclosporin A. Cellular localization of NF–κB was studied by confocal microscopy. Cyclosporin A and PSC-833 did not affect the cytoplasmic localization of NF–κB, whereas mitoxantrone translocated NF–κB to the nucleus, and this translocation was not affected by the presence of cyclosporin A.
Effect of cyclosporin A, PSC-833, and mitoxantrone on NF–κB expression levels and cellular localization. NF–κB expression levels, studied by flow cytometry, were not affected by exposure to cyclosporin A, PSC-833, mitoxantrone or the combination of mitoxantrone + cyclosporin A. Cellular localization of NF–κB was studied by confocal microscopy. Cyclosporin A and PSC-833 did not affect the cytoplasmic localization of NF–κB, whereas mitoxantrone translocated NF–κB to the nucleus, and this translocation was not affected by the presence of cyclosporin A.
Finally, nuclear/cytoplasmic distribution of doxorubicin was studied by confocal microscopy in 8226/MR20 cells, which coexpress LRP with BCRPR482, after incubation with and without each of the modulators cyclosporin A and PSC-833. Doxorubicin was used instead of mitoxantrone because, unlike mitoxantrone, it is not a substrate for BCRP R482, and modulation of intracellular transport could therefore be studied without a confounding effect on cellular retention. There was a marked increase in the intranuclear content of doxorubicin in the presence of cyclosporin A, as compared with the presence of PSC-833 or the absence of either modulator (Fig. 3A). In addition, cyclosporin A significantly (P < 0.05) decreased the IC50 of doxorubicin in 8226/MR20 cells, whereas neither PSC nor fumitremorgin C altered doxorubicin cytotoxicity (Fig. 3B). The effect of cyclosporin A on doxorubicin toxicity occurred in the absence of an effect on doxorubicin uptake (Fig. 3C), as expected, because doxorubicin is not a substrate for BCRPR482. The effect of cyclosporin A on nuclear-cytoplasmic drug distribution was confirmed in a second cell line that expresses LRP, HT1080/DR4. PSC-833 had no effect on nuclear-cytoplasmic distribution of doxorubicin in HT1080/DR4 cells either by itself or when MK571 was also present to ensure optimal cellular drug uptake, because HT1080/DR4 cells coexpress MRP-1. Finally, neither cyclosporin A nor PSC-833 had an effect on nuclear-cytoplasmic doxorubicin distribution in parental 8226/S or HT1080 cells.
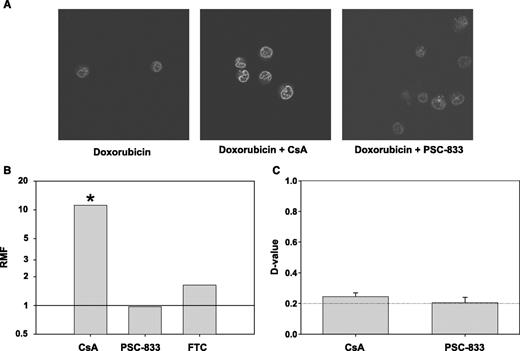
A, modulation of nuclear-cytoplasmic distribution of doxorubicin by cyclosporin A in 8226/MR20 cells, which overexpress LRP. 8226/MR20 cells were studied by confocal microscopy after incubation with doxorubicin alone and doxorubicin in the presence of cyclosporin A or PSC-833. B, cyclosporin A sensitizes 8226/MR20 cells to doxorubicin, whereas PSC-833 and fumitremorgin C (FTC) do not. Columns, geometric means of triplicate experiments. *, significant difference in RMF (P < 0.05) between a modulated cell line and RMF = 1. C, cyclosporin A has minimal effect on 8226/MR20 cell uptake of doxorubicin, consistent with the fact that 8226/MR20 cells express BCRPR482, for which doxorubicin is not a substrate. D values compare histograms measuring doxorubicin content after uptake with and without modulator. Columns, means of triplicate experiments; bars, SE.
A, modulation of nuclear-cytoplasmic distribution of doxorubicin by cyclosporin A in 8226/MR20 cells, which overexpress LRP. 8226/MR20 cells were studied by confocal microscopy after incubation with doxorubicin alone and doxorubicin in the presence of cyclosporin A or PSC-833. B, cyclosporin A sensitizes 8226/MR20 cells to doxorubicin, whereas PSC-833 and fumitremorgin C (FTC) do not. Columns, geometric means of triplicate experiments. *, significant difference in RMF (P < 0.05) between a modulated cell line and RMF = 1. C, cyclosporin A has minimal effect on 8226/MR20 cell uptake of doxorubicin, consistent with the fact that 8226/MR20 cells express BCRPR482, for which doxorubicin is not a substrate. D values compare histograms measuring doxorubicin content after uptake with and without modulator. Columns, means of triplicate experiments; bars, SE.
DISCUSSION
We have shown that cyclosporin A is a broad-spectrum MDR modulator, with effects on the MDR-associated ABC proteins that are expressed in AML and other malignancies, including Pgp, MRP-1, and BCRPR482. In addition, cyclosporin A also modulates BCRPR482T and BCRPR482G. In contrast, whereas PSC-833 effectively modulates Pgp, it has no effects on MRP-1, BCRPR482, BCRPR482T, or BCRPR482G. Finally, cyclosporin A enhanced nuclear doxorubicin uptake in cell lines that overexpressed LRP, whereas PSC-833 did not. Thus, cyclosporin A is a broad-spectrum MDR modulator with activity against Pgp, MRP-1, BCRP, and LRP.
In relation to ABC protein–specific modulators, broad-spectrum MDR modulators have the obvious advantage of overcoming multiple resistance mechanisms that may be present in malignant cells. In addition, modulation as part of initial therapy may prevent subsequent emergence of ABC protein–mediated resistance (28, 29), and early broad-spectrum modulation might therefore prevent subsequent expression of multiple ABC proteins. A potential disadvantage of broad-spectrum modulators is a lesser degree of efficacy against individual MDR proteins. The RMFs of cyclosporin A in cells expressing MRP-1 and BCRPR482 were one third to one half those of MK571 and fumitremorgin C, respectively, but are still likely to be sufficient to be associated with clinical sensitization. The efficacy of broad-spectrum modulation has yet to undergo systematic clinical testing.
In addition to the current findings with respect to cyclosporin A, we have previously shown that the clinically applicable MDR modulator VX-710 (biricodar) is effective against Pgp, MRP-1, and BCRPR482, but not BCRPR482T or BCRPR482G (20). We have also found that the taxane-based reversal agent tRA 98006 is effective against Pgp, MRP-1, BCRPR482, BCRPR482T, and BCRPR482G (19). VX-710 has no effect on nuclear-cytoplasmic drug transport (20), whereas the effect of tRA 98006 has not been studied. VX-710 is in clinical trials, whereas tRA 98006 has not yet undergone preclinical testing.
LRP has been found to be clinically significant in AML (14). The only previously characterized in vitro modulator of LRP is PAK-104P (30), which was not developed for clinical application. Cyclosporin A was not previously known to modulate LRP, and other clinical LRP modulators remain to be identified and characterized.
Both specific and broad-spectrum modulators have theoretical advantages for clinical MDR modulation. Clinical MDR seems to be multifactorial, and effective modulation may require targeting of multiple transport proteins. The use of a single broad-spectrum modulator may be preferable to the use of combinations of specific modulators to prevent drug interactions and cumulative toxicities. On the other hand, the use of specific modulators might minimize unwanted modulation of nontargeted transporters and minimize effects on nontumor tissues such as normal hematopoietic stem cells, bile canaliculi, and the blood-brain barrier. Clinical application of broad-spectrum modulation warrants testing in clinical trials with correlative laboratory studies.
In addition to broad-spectrum modulation, cyclosporin A has been reported to have other effects that may be beneficial. These include induction of apoptosis in at least some cell types (31, 32) as well as antiangiogenic effects (33). Enhancement of the apoptotic response by cyclosporin A has been attributed to inhibition of activation of NF–κB (31). However, we found that neither cyclosporin A nor PSC-833 affected the expression level or nuclear translocation of the NF–κB p65 subunit in HL60 cells, nor did cyclosporin A affect the activation of NF–κB associated with mitoxantrone exposure. Thus, the effects of cyclosporin A on NF–κB activation may be cell type– and/or NF–κB activation trigger–dependent. The relative importance of broad-spectrum modulation and other effects of cyclosporin A to its shown clinical efficacy in AML remains to be determined.
Broad-spectrum modulation by cyclosporin A may make it effective in diverse malignancies in which multiple MDR proteins are expressed. In addition to the positive clinical trial in AML (5), clinical trials of cyclosporin A in combination with chemotherapy regimens in solid tumors have also generated encouraging results (34–38).
Grant support: National Cancer Institute grant R21 CA 98457, a Leukemia and Lymphoma Society Translational Research Program grant, NIH grant T32 CA09072-28, shared resources of the Roswell Park Cancer Center Support grant P30 CA16056, the Leonard S. LoVullo Memorial Fund for Leukemia Research, and the Dennis J. Szefel Jr. Endowed Fund for Leukemia Research at Roswell Park Cancer Institute.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.