


Abstract
The oncogenic MUC1-C protein is overexpressed in triple-negative breast cancer (TNBC) cells and contributes to their epigenetic reprogramming and chemoresistance. Here we show that targeting MUC1-C genetically or pharmacologically with the GO-203 inhibitor, which blocks MUC1-C nuclear localization, induced DNA double-strand breaks and potentiated cisplatin (CDDP)-induced DNA damage and death. MUC1-C regulated nuclear localization of the polycomb group proteins BMI1 and EZH2, which formed complexes with PARP1 during the DNA damage response. Targeting MUC1-C downregulated BMI1-induced H2A ubiquitylation, EZH2-driven H3K27 trimethylation, and activation of PARP1. As a result, treatment with GO-203 synergistically sensitized both mutant and wild-type BRCA1 TNBC cells to the PARP inhibitor olaparib. These findings uncover a role for MUC1-C in the regulation of PARP1 and identify a therapeutic strategy for enhancing the effectiveness of PARP inhibitors against TNBC.
These findings demonstrate that targeting MUC1-C disrupts epigenetics of the PARP1 complex, inhibits PARP1 activity, and is synergistic with olaparib in TNBC cells.
Introduction
MUC1-C is a transmembrane oncoprotein that is aberrantly overexpressed by diverse types of human carcinomas (1, 2). MUC1-C interacts with receptor tyrosine kinases, such as HER2 and EGFR, at the cell membrane and promotes their activation and downstream signaling pathways (2–4). MUC1-C is also imported into the nucleus of cancer cells, where it induces changes in gene expression patterns by direct interactions with transcription factors and effectors of epigenetic regulation (3, 4). MUC1-C–induced gene signatures in tumors have been associated with poor clinical outcomes and multiple hallmarks of the cancer cell, including the epithelial–mesenchymal transition (EMT), cancer stem cell (CSC) state, repression of tumor suppressor genes (TSG), reprogramming of the epigenome and immune evasion (3, 4). EMT and the CSC state contribute to the development of anticancer drug resistance (5, 6). In this respect, MUC1-C promotes resistance to genotoxic anticancer agents (7–11). Studies have shown that MUC1-C blocks the apoptotic response of cancer cells to cisplatin (CDDP) and etoposide treatment (7–9). In addition, resistance to gemcitabine has been associated with MUC1-C–induced increases in anabolic glucose metabolism (11). These findings supported the notion that MUC1-C activates pleotropic cellular mechanisms that confer resistance to genotoxic anticancer agents.
An early event in the response to genotoxic stress is the recruitment and activation of PARP1 at sites of DNA lesions (12). PARP1 catalyzes the poly-ADP-ribosylation (PARylation) of itself and multiple target proteins in initiating the repair of single-strand and double-strand breaks (DSB; ref. 12). In the repair of DSBs by homologous recombination (HR), PARP1 functions in the recruitment of DNA damage response (DDR) proteins, such as phosphorylated histone H2AX (γH2AX; ref. 12). PARP1 also recruits the breast cancer type 1 susceptibility protein (BRCA1), which induces DSB resection and loading of RAD51 for strand exchange in HR (13, 14). PARP1 ADP-ribosylates BRCA1, resulting in decreased affinity of BRCA1 for DNA binding and fine-tuning of its function in HR repair (15). The importance of PARP1 in the DDR led to the development of selective inhibitors, such as olaparib, rucaparib, and niraparib, for the treatment of tumors with BRCA1/BRCA2 mutations or deletions (16). Trapping of PARP function by inhibitors with the resulting generation of replication-dependent DSBs contributes to the synthetic lethal relationship between PARP and BRCA (17). In addition, the synergy between PARP inhibition and treatment with genotoxic anticancer agents has provided the basis for evaluating such combinations in clinical trials (18, 19). Notably, PARP1 also plays roles in (i) transcriptional regulation of activated genes, (ii) maintaining the stability of replication forks, and (iii) remodeling of chromatin structure (12, 20). In this way, PARP1 inhibition in cancer cells can affect multiple pathways, including those associated with the acquisition of drug resistance.
These studies uncover a previously unrecognized role for MUC1-C in the DDR. We show that targeting MUC1-C genetically or pharmacologically with the cell-penetrating GO-203 inhibitor (21) suppresses nuclear BMI1 and EZH2 levels, and thereby their chromatin remodeling activities, which are necessary for DSB-mediated transcriptional silencing and DNA repair. Additionally, targeting MUC1-C suppresses activation of PARP1 in the DDR and is synergistic with the PARP inhibitor olaparib in the treatment of triple-negative breast cancer (TNBC) cells with mutant and wild-type BRCA1.
Materials and Methods
Cell culture
Human BT-549 (wt BRCA1) TNBC cells were grown in RPMI1640 medium (Corning) supplemented with 10% FBS, 100 μg/mL streptomycin, 100 U/mL penicillin, and 10 μg/mL insulin. SUM149 (mut BRCA1) and SUM159 (wt BRCA1) TNBC cells were cultured in Ham's F-12 medium (Corning) supplemented with 10 mmol/L HEPES, 5% FBS, 100 μg/mL streptomycin, 100 U/mL penicillin, 5 μg/mL insulin, and 1 μg/mL hydrocortisone. MDA-MB-468 (wt BRCA1) were grown in DMEM containing 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin. BT-549 and SUM149 cells transduced to stably express a tet-CshRNA or a tet-MUC1shRNA were treated with doxycycline (DOX; Sigma). Cells were also treated with cisplatin (Santa Cruz Biotechnology), etoposide (Sigma), olaparib (Selleck Chemicals), and GO-203, which is a cell-penetrating peptide that blocks MUC1-C homodimerization, nuclear localization and oncogenic function (21).
Subcellular fractionation
Cells were washed with PBS and incubated in cell lysis buffer (10 mmol/L HEPES, pH 8.0, 1.5 mmol/L MgCl2, 0.5% NP40, and 10 mmol/L KCl) for 10 minutes at 4°C. The total cell lysates were centrifuged at 4,000 rpm for 5 minutes at 4°C and the pellets were incubated in nuclear lysis buffer (10 mmol/L HEPES pH 8.0, 1.6 mmol/L MgCl2, 0.5% NP40, 420 mmol/L NaCl, 0.2 mmol/L EDTA, and 25% glycerol) for 20 minutes at 4°C, and then sheared by passage through 20 to 26 gauge needles. After centrifugation at 13,000 rpm for 10 minutes, the supernatants were collected as nuclear lysates.
Immunoblot analysis
Lysates were subjected to immunoblotting with anti-MUC1-C (HM-1630-P1ABX; ThermoFisher Scientific), anti-β-actin (A5441; Sigma), anti-lamin A/C (4777), anti-γH2AX (9718), anti-ATR (2790), anti-pCHK1 (2348), anti-CHK1 (2360), anti-pBRCA1 (9009), anti-BRCA1 (14823), anti-BMI1 (6964), anti-H2AUb1 (8240), anti-H2A (12349), anti-EZH2 (5246), anti-H3K27me3 (9733), anti-H3 (9715), anti-H3K56ac (4243), anti-PARP1 (9532) (Cell Signaling Technology), anti-pATR (GTX128145; GeneTex), and anti-FANCD2 (ac108928; Abcam). Signal intensity was determined using ImageJ 1.51k software (NIH).
Confocal microscopy
Cells were fixed in 4% paraformaldehyde at room temperature for 20 minutes. The samples were washed three times with PBS and then incubated with 0.3% Triton X-100 (Sigma) at room temperature for 15 minutes. Samples were next blocked with 3% BSA and incubated with primary antibodies at 4°C. The samples were then incubated with goat anti-Armenian hamster IgG H&L labeled with Alexa Fluor 488 (Abcam) and goat anti-rabbit IgG H&L labeled with Alexa Fluor 568 (Abcam) at room temperature for 60 minutes. DAPI (ThermoFisher Scientific) was used for staining of nuclei. The cells were analyzed by confocal microscopy using an inverted Leica TCS SP5 scope (Leica). Images were captured at increments and converted to composites localized on the nucleus by Leica LAS AF software (22).
Colony formation assay
Cells were seeded at 3,000 cells/well in six-well plates and treated with 500 ng/mL DOX and/or 10 μmol/L CDDP for 10 days. The cells were then stained with 0.5% crystal violet in 25% methanol. Colonies >25 cells were counted in triplicate wells.
Cell viability and combination index
Cells were seeded at a density of 1,000 to 2,000 cells per well in 96-well plates. After 24 hours, the cells were treated with different concentrations of GO-203, CDDP and olaparib alone and in combination. Cell viability was assessed by Alamar blue staining of triplicate wells. The IC50 value was determined by nonlinear regression of the dose–response data using Prism 7.0 (GraphPad Software). Drug interaction and dose–effect relationships were analyzed according to reported methods (23). The combination index (CI) was calculated to assess synergism (CI < 1) or antagonism (CI > 1).
Coimmunoprecipitation of nuclear proteins
Nuclear lysates were isolated as described above. DNA was digested by incubation in 20 U/mL DNase for 30 minutes at 37°C. Nuclear proteins were incubated with primary antibodies at 4°C overnight and then precipitated with Dynabeads Protein G (10003D; ThermoFisher Scientific) for 2 hours at 4°C. Beads were washed twice with washing buffer (20 mmol/L Tris-HCl, pH 8.0, 0.2 mmol/L EDTA, 1.5 mmol/L MgCl2, 0.5% NP40, and 150 mmol/L NaCl) and once with 10% TE buffer (BM-304A; Boston BioProducts), and then resuspended in sample loading buffer.
Measurement of PARP1 activity
The PARP Activity Kit (4685-096-K; R&D Systems) was used to measure PARP activity according to the manufacturer's instructions.
Tumor model studies
Six- to 8-week-old female nude mice (Taconic Farm) were injected subcutaneously in the flank with 3 × 106 SUM149 cells in 50% matrigel solution. Mice were grouped when tumor size reached approximately 100 mm3 and were treated intraperitoneally with vehicle, 15 mpk GO-203 encapsulated in nanoparticles (GO-203/NPs) for sustained delivery (once a week for three weeks; ref. 21), 15 mpk olaparib (5 days on, 2 days off for three weeks), and GO-203/NPs plus olaparib. Tumor measurements and body weights were recorded twice a week. Tumor volumes were calculated by the formula: (width)2 × length/2.
Animals
Animal studies were performed under animal protocol #03-029 approved by the Dana-Farber Cancer Institute Animal Use and Care Committee.
Statistical analysis
Each experiment was repeated at least three times. Data are expressed as the mean ± SD. The unpaired Student t test was used to examine differences between means of two groups. A P-value of <0.05 was considered a statistically significant difference denoted by an asterisk.
Results
MUC1-C localizes to the nucleus in the response to DNA damage
MUC1-C consists of a 58 aa extracellular domain, a 22 aa transmembrane domain, and a 72 aa cytoplasmic domain (Fig. 1A). The MUC1-C cytoplasmic domain is an intrinsically disordered protein, which includes a CQC motif necessary for MUC1-C homodimerization and nuclear import (24–26). MUC1-C is constitutively expressed in the nucleus of diverse cancer cells (2, 4, 24, 27); however, its nuclear localization has not been associated with DNA damage. We found that treatment of BT-549 TNBC cells with CDDP has little effect on total MUC1-C levels (Fig. 1B, left), but is associated with increases in nuclear MUC1-C protein (Fig. 1B, right). Similar results were obtained in the response of MDA-MB-468 (Supplementary Fig. S1A), SUM159 (Supplementary Fig. S1B), and BRCA1 mutant SUM149 (Fig. 1C) TNBC cells to CDDP treatment. As confirmation of these observations, confocal microscopy further showed CDDP-induced localization of MUC1-C to the nucleus in BT-549 and SUM149 cells (Fig. 1D and E). Similar effects were observed with the genotoxic agent, etoposide (Supplementary Fig. S1C and S1D), indicating that MUC1-C is imported to the nucleus in the response to DNA damage.
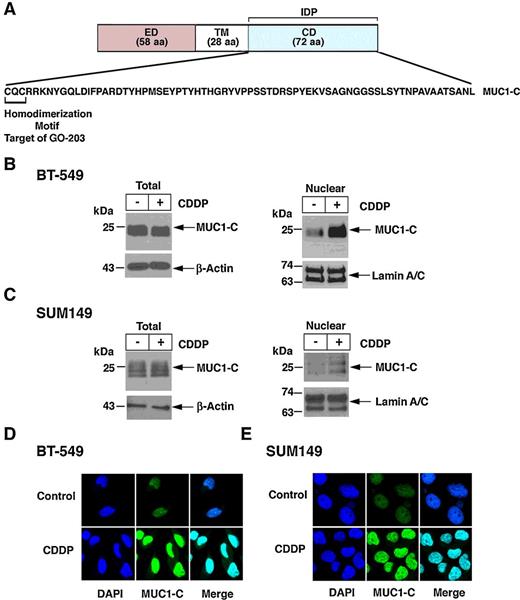
Nuclear localization of MUC1-C in the DDR. A, Structure of the MUC1-C protein with the 58 aa extracellular domain (ED), 28 aa transmembrane domain (TM), and 72 aa cytoplasmic domain (CD). Highlighted is the aa sequence of the CD, which is an intrinsically protein (IDP). The CQC motif is necessary for MUC1-C homodimerization and nuclear localization, and is the target of the cell-penetrating peptide GO-203 (R9-CQCRRKN). B and C, BT-549 (B) and SUM149 (C) cells were left untreated or treated with 10 μmol/L CDDP for 24 hours. Total cell (left) and nuclear (right) lysates were analyzed by immunoblotting with antibodies against the indicated proteins. D and E, BT-549 (D) and SUM149 (E) cells were left untreated (Control) or treated with 10 μmol/L CDDP for 24 hours. The cells were stained with DAPI and analyzed for nuclear MUC1-C localization by confocal microscopy.
Nuclear localization of MUC1-C in the DDR. A, Structure of the MUC1-C protein with the 58 aa extracellular domain (ED), 28 aa transmembrane domain (TM), and 72 aa cytoplasmic domain (CD). Highlighted is the aa sequence of the CD, which is an intrinsically protein (IDP). The CQC motif is necessary for MUC1-C homodimerization and nuclear localization, and is the target of the cell-penetrating peptide GO-203 (R9-CQCRRKN). B and C, BT-549 (B) and SUM149 (C) cells were left untreated or treated with 10 μmol/L CDDP for 24 hours. Total cell (left) and nuclear (right) lysates were analyzed by immunoblotting with antibodies against the indicated proteins. D and E, BT-549 (D) and SUM149 (E) cells were left untreated (Control) or treated with 10 μmol/L CDDP for 24 hours. The cells were stained with DAPI and analyzed for nuclear MUC1-C localization by confocal microscopy.
Targeting MUC1-C induces γH2AX and sensitizes to CDDP treatment
To determine if MUC1-C is functionally involved in the DDR, we generated BT-549 cells expressing a tetracycline-inducible control shRNA (tet-CshRNA) or a MUC1-C shRNA (tet-MUC1shRNA). DOX treatment of BT-549/tet-MUC1shRNA, but not BT-549/tet-CshRNA, cells was associated with downregulation of MUC1-C and, interestingly, increases in γH2AX (Fig. 2A; Supplementary Fig. S2A). Silencing MUC1-C in SUM149 cells also resulted in induction of γH2AX (Fig. 2B; Supplementary Fig. S2B), indicating that downregulation of MUC1-C induces DSBs. In concert with these observations, we found that silencing MUC1-C increases CDDP-induced γH2AX levels (Fig. 2C and D). Silencing MUC1-C also potentiated CDDP-induced loss of colony formation (Fig. 2E and F), consistent with a role for MUC1-C in DNA damage-induced cell death.

Silencing MUC1-C induces γH2AX. A and B, BT-549 (A) and SUM149 (B) cells stably expressing a tet-MUC1shRNA were treated with 500 ng/mL DOX for 72 hours. Total cell lysates were immunoblotted with antibodies against the indicated proteins. C and D, BT-549/tet-MUC1shRNA (C) and SUM149/tet-MUC1shRNA (D) cells were treated with 500 ng/mL DOX for 72 hours and then 10 μmol/L CDDP for 24 hours. Lysates were subjected to immunoblot analysis. E and F, BT-549/tet-MUC1shRNA (E) and SUM149/tet-MUC1shRNA (F) cells were treated with 500 ng/mL DOX and/or 10 μmol/L CDDP for 10 days. Colony formation was determined by crystal violet staining. The results are expressed as the mean ± SD of three determinations. *, P < 0.05.
Silencing MUC1-C induces γH2AX. A and B, BT-549 (A) and SUM149 (B) cells stably expressing a tet-MUC1shRNA were treated with 500 ng/mL DOX for 72 hours. Total cell lysates were immunoblotted with antibodies against the indicated proteins. C and D, BT-549/tet-MUC1shRNA (C) and SUM149/tet-MUC1shRNA (D) cells were treated with 500 ng/mL DOX for 72 hours and then 10 μmol/L CDDP for 24 hours. Lysates were subjected to immunoblot analysis. E and F, BT-549/tet-MUC1shRNA (E) and SUM149/tet-MUC1shRNA (F) cells were treated with 500 ng/mL DOX and/or 10 μmol/L CDDP for 10 days. Colony formation was determined by crystal violet staining. The results are expressed as the mean ± SD of three determinations. *, P < 0.05.
To extend these studies, cells were treated with the GO-203 inhibitor, which targets the MUC1-C cytoplasmic domain at a CQC motif required for MUC1-C homodimerization and nuclear import (Fig. 1A; refs. 2, 24). Treatment of BT-549 and SUM149 cells with GO-203 had little effect on MUC1-C levels (Supplementary Fig. S3A and S3B), but attenuated CDDP-induced localization of MUC1-C to the nucleus (Fig. 3A and B). Notably and consistent with the effects of silencing MUC1-C, treatment with GO-203 alone and in combination with CDDP was associated with increases in γH2AX levels (Fig. 3C and D). These effects of GO-203 on MUC1-C localization and induction of γH2AX were confirmed by confocal microscopy (Fig. 3E and F). In concert with these observations, we found that GO-203 is synergistic with CDDP in inducing cell death (Fig. 3G and H; Supplementary Fig. S4A and S4B). These findings demonstrated that targeting MUC1-C genetically or pharmacologically increases γH2AX and potentiates CDDP-induced loss of TNBC cell survival.
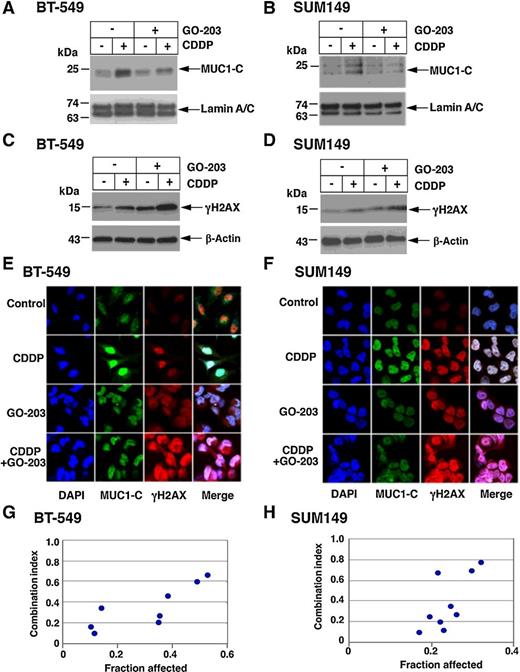
Targeting MUC1-C with the GO-203 inhibitor attenuates nuclear localization of MUC1-C and induces γH2AX. A–D, BT-549 (A and C) and SUM149 (B and D) cells were left untreated or treated with 1 μmol/L GO-203 for 48 hours and then without or with 10 μmol/L CDDP for 24 hours. Nuclear (A and B) and total (C and D) lysates were immunoblotted with antibodies against the indicated proteins. E and F, BT-549 (E) and SUM149 (F) cells were treated with 10 μmol/L CDDP for 24 hours, 1 μmol/L GO-203 for 48 hours, or GO-203 for 48 hours and then CDDP for 24 hours. The cells were stained with DAPI and analyzed for nuclear MUC1-C and γH2AX by confocal microscopy. G and H, BT-549 (G) and SUM149 (H) cells were left untreated or treated with 0.1, 0.3, or 1 μmol/L GO-203 in the absence and presence of different concentrations of CDDP (Supplementary Fig. S4A and S4B). Cell viability was determined by Alamar blue staining. The results represent the CI values, which at <1.0 denote synergism.
Targeting MUC1-C with the GO-203 inhibitor attenuates nuclear localization of MUC1-C and induces γH2AX. A–D, BT-549 (A and C) and SUM149 (B and D) cells were left untreated or treated with 1 μmol/L GO-203 for 48 hours and then without or with 10 μmol/L CDDP for 24 hours. Nuclear (A and B) and total (C and D) lysates were immunoblotted with antibodies against the indicated proteins. E and F, BT-549 (E) and SUM149 (F) cells were treated with 10 μmol/L CDDP for 24 hours, 1 μmol/L GO-203 for 48 hours, or GO-203 for 48 hours and then CDDP for 24 hours. The cells were stained with DAPI and analyzed for nuclear MUC1-C and γH2AX by confocal microscopy. G and H, BT-549 (G) and SUM149 (H) cells were left untreated or treated with 0.1, 0.3, or 1 μmol/L GO-203 in the absence and presence of different concentrations of CDDP (Supplementary Fig. S4A and S4B). Cell viability was determined by Alamar blue staining. The results represent the CI values, which at <1.0 denote synergism.
To assess whether targeting MUC1-C alters DNA damage checkpoints, we investigated effects on activation of ATR and CHK1. Silencing MUC1-C in BT-549 (Supplementary Fig. S5A) and SUM149 (Supplementary Fig. S5B) cells was associated with increases in pATR and pCHK1 in the absence of changes in ATR and CHK1 levels. Similar responses were observed when BT-549 (Supplementary Fig. S5C) and SUM149 (Supplementary Fig. S5D) cells were treated with GO-203, indicating that MUC1-C downregulation or inhibition activates the ATR and CHK1 checkpoints. We also found that targeting MUC1-C increases BRCA1 phosphorylation and has little if any effect on FANCD2 levels (Supplementary Fig. S5A–S5D). These results provided support for a model in which MUC1-C induces DSBs by mechanisms other than affecting these DNA damage checkpoints or repair proteins.
MUC1-C regulates nuclear BMI1 in the DDR
Nuclear functions of MUC1-C have been linked to the repression of TSGs and reprogramming of the epigenome by regulating polycomb repressive complex 1 (PRC1) function (3, 4). Here, we found that MUC1-C forms a nuclear complex with BMI1 (Fig. 4A and B) and that silencing MUC1-C is associated with decreases in nuclear BMI1 (Fig. 4C and D). BMI1 regulates the DDR by ubiquitylation of H2A and thereby facilitating the repair of DSBs by HR and NHEJ (28). In this capacity, we found that downregulation of MUC1-C decreases nuclear H2AUb1 levels (Fig. 4C and D). Consistent with these results, we also found that targeting MUC1-C with the GO-203 inhibitor disrupts the interaction between MUC1-C and BMI1 (Fig. 4E and F). Moreover, inhibiting MUC1-C decreased nuclear BMI1 expression and in turn markedly reduced nuclear H2AUb1 levels (Fig. 4G and H), indicating that MUC1-C contributes to BMI1 function in the DDR.

Targeting MUC1-C downregulates nuclear BMI1 and H2AUb1 in the DDR. A and B, Nuclear proteins from BT-549 (A) and SUM149 (B) cells were precipitated with anti-MUC1-C or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. C and D, BT-549/tet-MUC1shRNA (C) and SUM149/tet-MUC1shRNA (D) cells were treated with 500 ng/mL DOX for 72 hours and then 10 μmol/L CDDP for 24 hours. Nuclear lysates were immunoblotted with antibodies against the indicated proteins. E and F, BT-549 (E) and SUM149 (F) cells were left untreated or treated with 3 μmol/L GO-203 for 48 hours. Nuclear proteins were precipitated with anti-MUC1-C or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. G and H, BT-549 (G) and SUM149 (H) cells were left untreated or treated with 3 μmol/L GO-203 for 48 hours. Nuclear lysates were analyzed by immunoblotting with antibodies against the indicated proteins.
Targeting MUC1-C downregulates nuclear BMI1 and H2AUb1 in the DDR. A and B, Nuclear proteins from BT-549 (A) and SUM149 (B) cells were precipitated with anti-MUC1-C or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. C and D, BT-549/tet-MUC1shRNA (C) and SUM149/tet-MUC1shRNA (D) cells were treated with 500 ng/mL DOX for 72 hours and then 10 μmol/L CDDP for 24 hours. Nuclear lysates were immunoblotted with antibodies against the indicated proteins. E and F, BT-549 (E) and SUM149 (F) cells were left untreated or treated with 3 μmol/L GO-203 for 48 hours. Nuclear proteins were precipitated with anti-MUC1-C or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. G and H, BT-549 (G) and SUM149 (H) cells were left untreated or treated with 3 μmol/L GO-203 for 48 hours. Nuclear lysates were analyzed by immunoblotting with antibodies against the indicated proteins.
MUC1-C interacts with EZH2 in the DDR
MUC1-C also plays a role in regulating EZH2 (29), which induces H3K27 trimethylation (H3K27me3) and has been linked to the DDR (30). In this respect, we found that MUC1-C forms nuclear complexes with EZH2 (Fig. 5A and B) and that silencing MUC1-C decreases nuclear EZH2 in association with suppression of H3K27me3 levels (Fig. 5C and D). Inhibiting MUC1-C with GO-203 also decreased (i) the interaction between nuclear MUC1-C and EZH2 (Fig. 5E and F), and (ii) nuclear EZH2 and H3K27me3 levels (Fig. 5G and H). In contrast, targeting MUC1-C had no apparent effect on H3K56ac levels (Supplementary Fig. S6A–S6D), which have been implicated in DNA repair (31). These findings demonstrate that, in addition to BMI1, MUC1-C regulates nuclear localization of EZH2 and its function in catalyzing the formation of H3K27me3, which is necessary for the repair of DSBs.

MUC1-C forms a nuclear complex with EZH2 and regulates H3K27 trimethylation. A and B, Nuclear proteins from BT-549 (A) and SUM149 (B) cells were precipitated with anti-MUC1-C or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. C and D, BT-549/tet-MUC1shRNA (C) and SUM149/tet-MUC1shRNA (D) cells were treated with 500 ng/mL DOX for 72 hours and then 10 μmol/L CDDP for 24 hours. Nuclear lysates were immunoblotted with antibodies against the indicated proteins. E and F, BT-549 (E) and SUM149 (F) cells were left untreated or treated with 3 μmol/L GO-203 for 48 hours. Nuclear proteins were precipitated with anti-MUC1-C or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. G and H, BT-549 (G) and SUM149 (H) cells were left untreated or treated with 3 μmol/L GO-203 for 48 hours. Nuclear lysates were analyzed by immunoblotting with antibodies against the indicated proteins.
MUC1-C forms a nuclear complex with EZH2 and regulates H3K27 trimethylation. A and B, Nuclear proteins from BT-549 (A) and SUM149 (B) cells were precipitated with anti-MUC1-C or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. C and D, BT-549/tet-MUC1shRNA (C) and SUM149/tet-MUC1shRNA (D) cells were treated with 500 ng/mL DOX for 72 hours and then 10 μmol/L CDDP for 24 hours. Nuclear lysates were immunoblotted with antibodies against the indicated proteins. E and F, BT-549 (E) and SUM149 (F) cells were left untreated or treated with 3 μmol/L GO-203 for 48 hours. Nuclear proteins were precipitated with anti-MUC1-C or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. G and H, BT-549 (G) and SUM149 (H) cells were left untreated or treated with 3 μmol/L GO-203 for 48 hours. Nuclear lysates were analyzed by immunoblotting with antibodies against the indicated proteins.
MUC1-C forms a complex with PARP1 and regulates PARP activity
PARP1 plays multiple roles in DNA repair, including the modulation of chromatin structure and repair of DSBs by recruitment of BMI1 and EZH2 to sites of DNA damage (12, 28, 30, 32, 33). As reported for BMI1 and EZH2, MUC1-C was also detectable in nuclear complexes with PARP1 (Fig. 6A; Supplementary Fig. S7A). The formation of nuclear MUC1-C/PARP1 complexes was increased by CDDP treatment (Fig. 6B and C), but was unaffected by olaparib exposure (Fig. 6D; Supplementary Fig. S5B), indicating that the association is independent of PARP-mediated PARylation. Targeting MUC1-C had no apparent effect on PARP1 expression (Supplementary Fig. S7C and S7D). However, targeting MUC1-C with silencing or GO-203 treatment significantly decreased PARP1 activity when used alone and in combination with olaparib (Fig. 6E and F), demonstrating that MUC1-C is of importance for PARP1 activation.

MUC1-C forms a complex with PARP1 and regulates PARP activity. A–D, BT-549 cells were left untreated and treated with (i) 10 μmol/L CDDP for 24 hours (A), (ii) 10 μmol/L CDDP for the indicated times (B and C), or (iii) 3 μmol/L olaparib (D) for 48 hours. Nuclear proteins were precipitated with anti-MUC1-C, anti-PARP1 or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. Intensity of the signals in B and C (top) were determined using ImageJ 1.51k software and are expressed as relative values compared with that obtained for untreated cells (assigned a value of 1). The results are representative of two separate experiments. E and F, BT-549/tet-MUC1shRNA (E) and SUM149/tet-MUC1shRNA (F) cells were treated with 500 ng/mL DOX, 1 μmol/L GO-203, or 3 μmol/L olaparib alone and in combination for 7 days. Lysates were analyzed for PARP activity. The results are expressed as relative PARP activity (mean ± SD of three determinations) as compared with that obtained from control cells (assigned a value of 1). *, P < 0.05.
MUC1-C forms a complex with PARP1 and regulates PARP activity. A–D, BT-549 cells were left untreated and treated with (i) 10 μmol/L CDDP for 24 hours (A), (ii) 10 μmol/L CDDP for the indicated times (B and C), or (iii) 3 μmol/L olaparib (D) for 48 hours. Nuclear proteins were precipitated with anti-MUC1-C, anti-PARP1 or a control IgG. Input and the precipitates were analyzed by immunoblotting with antibodies against the indicated proteins. Intensity of the signals in B and C (top) were determined using ImageJ 1.51k software and are expressed as relative values compared with that obtained for untreated cells (assigned a value of 1). The results are representative of two separate experiments. E and F, BT-549/tet-MUC1shRNA (E) and SUM149/tet-MUC1shRNA (F) cells were treated with 500 ng/mL DOX, 1 μmol/L GO-203, or 3 μmol/L olaparib alone and in combination for 7 days. Lysates were analyzed for PARP activity. The results are expressed as relative PARP activity (mean ± SD of three determinations) as compared with that obtained from control cells (assigned a value of 1). *, P < 0.05.
Olaparib and GO-203 are synergistic in the treatment of TNBC cells
The demonstration that MUC1-C contributes to PARP1 activity invoked the possibility that targeting MUC1-C could have an effect on olaparib sensitivity. To address this notion, we first defined the half-maximal inhibitory concentrations of olaparib and GO-203 when used alone (Supplementary Fig. S8A and S8B). In studies of BT-549 cells, low μmol/L concentrations of GO-203 were combined with olaparib (0.1–10 μmol/L; Fig. 7A), demonstrating synergistic activity with the combinations as evidenced by CI values of less than 1.0 (Fig. 7B). Similar results were obtained with SUM149 cells (Fig. 7C and D), indicating that the synergy occurs in TNBC cells with mutant and wild-type BRCA1. To extend these results, we also defined sensitivity of the wild-type BRCA1 MDA-MB-468 and SUM159 cells to olaparib and GO-203 alone (Supplementary Fig. S9A and S9B) and in combination, here again observing marked synergy for treatment with these agents (Supplementary Fig. S10A–S10D). The reproducibility of these results in four different types of TNBC cells provided strong support for the observed mechanistic interaction between these agents. Consistent with these findings, treatment of established SUM149 tumor xenografts with the combination of olaparib and GO-203 encapsulated in nanoparticles (GO-203/NPs) for sustained delivery was more effective in inhibiting growth than either agent alone (Fig. 7E) without an increase in toxicity (Supplementary Fig. S11). In concert with the results obtained in in vitro studies, analysis of tumors harvested on day 7 of treatment demonstrated decreases in MUC1-C and increases in γH2AX as compared with the control untreated cells (Fig. 7F). In contrast, analysis of tumors surviving GO-203/NP treatment alone and in combination with olaparib exhibited increases in MUC1-C levels and decreases in γH2AX (Fig. 7G), consistent in part with a role for MUC1-C in attenuating the formation of DSBs and blocking death of cancer cells in the DDR (7–9). These findings thus confirmed proof-of-mechanism observed in vitro and demonstrated that GO-203 can be combined with olaparib to enhance antitumor activity.

Synergistic activity of GO-203 and olaparib in the treatment of TNBC cells. A–D, BT-549 (A and B) and SUM149 (C and D) cells were left untreated or treated with 0.1, 0.3, or 1.0 μmol/L GO-203 in the absence or presence of the indicated concentrations of olaparib. Cell viability (mean ± SD of at least three determinations) was determined by Alamar blue staining (left) and was used to calculate the CI values (right), which at <1.0 denote synergism. E, Nude mice bearing established SUM149 tumors were randomized into four groups and treated intraperitoneally with vehicle control (blue squares), GO-203/NPs (green triangles), olaparib (orange diamonds), and GO-203/NPs plus olaparib (red circles). The results are expressed as tumor volume (mean ± SEM; six mice per group). F and G, Lysates from tumors harvested on day 7 (F) and at the end of treatment (G) were analyzed by immunoblotting with antibodies against the indicated proteins. *, P < 0.05.
Synergistic activity of GO-203 and olaparib in the treatment of TNBC cells. A–D, BT-549 (A and B) and SUM149 (C and D) cells were left untreated or treated with 0.1, 0.3, or 1.0 μmol/L GO-203 in the absence or presence of the indicated concentrations of olaparib. Cell viability (mean ± SD of at least three determinations) was determined by Alamar blue staining (left) and was used to calculate the CI values (right), which at <1.0 denote synergism. E, Nude mice bearing established SUM149 tumors were randomized into four groups and treated intraperitoneally with vehicle control (blue squares), GO-203/NPs (green triangles), olaparib (orange diamonds), and GO-203/NPs plus olaparib (red circles). The results are expressed as tumor volume (mean ± SEM; six mice per group). F and G, Lysates from tumors harvested on day 7 (F) and at the end of treatment (G) were analyzed by immunoblotting with antibodies against the indicated proteins. *, P < 0.05.
Discussion
Treatment of TNBC is limited by a lack of actionable targets and an aggressive phenotype that is often refractory to cytotoxic chemotherapeutic agents (34). MUC1-C is an oncoprotein that is expressed in over 90% of TNBCs (2, 35) and is of importance to multiple hallmarks of the TNBC cell, including EMT (36), the CSC state (37–39), and epigenetic reprogramming with downregulation of TSGs (29, 40, 41). In TNBCs, 6% to 8% harbor BRCA1 mutations and about 2.5% have BRCA2 mutations (42, 43). The present studies were undertaken to determine whether MUC1-C plays a role in DNA repair of TNBC cells. Unexpectedly, silencing of MUC1-C in settings of wild-type and mutant BRCA1 was associated with the induction of DSBs as detected by increases in γH2AX. Treatment with the MUC1-C inhibitor GO-203, which blocks MUC1-C function, also induced γH2AX, suggesting that MUC1-C may be involved in the repair of DSBs. In support of this notion, we found that treatment with CDDP, a DNA strand crosslinking agent widely used for TNBC therapy (34), is associated with increases in nuclear MUC1-C levels. Along these lines, similar results were obtained from treatment with etoposide, an inhibitor of topoisomerase II and inducer of DNA strand breaks, suggesting that MUC1-C is targeted to the nucleus in the response to DNA damage. In support of a potential role in the DDR, targeting MUC1-C in combination with CDDP resulted in more pronounced induction of DSBs than obtained with CDDP alone and synergistically enhanced CDDP-induced cytotoxicity. Notably, few insights are available regarding potential involvement of MUC1-C in the DDR. MUC1-C interacts directly with the ataxia–telangiectasia mutated (ATM) kinase and the ATM substrate H2AX in breast cancer cells (44). MUC1-C also promotes the repair of ionizing radiation (IR)-induced γH2AX foci (44). However, the mechanistic basis for these observations has remained unclear and is of potential importance for exploiting MUC1-C as a target for enhancing treatment of TNBCs with PARP inhibitors.
The repair of DNA lesions is dependent on the relaxation of chromatin structure and repression of transcription (45). In this respect, the repressive PcG proteins play important functional roles in the repair of DSBs. The PRC1 component BMI1 in association with the catalytic member RING2 ubiquitylate H2A on K119 in the response to DSBs (46, 47), a modification necessary for repressing the transcriptional machinery in the repair of DNA damage (48, 49). In this way, silencing BMI1 inhibits DSB repair and enhances sensitivity to DNA damage (46). We found that targeting MUC1-C decreases nuclear BMI1 levels and, significantly, suppresses H2AUb1 levels. In the hierarchical model, PRC1 is recruited to sites of PRC2-mediated H3K27 trimethylation (50, 51). Our results further show that targeting MUC1-C is associated with suppression of nuclear EZH2 and H3K27me3 levels, indicating that MUC1-C could contribute to DNA repair by activating PRC2 and thereby the recruitment of PRC1. In support of the importance of both complexes in DNA repair, PRC1 and PRC2 are recruited to sites of DNA damage and loss of either one decreases survival in the response to IR-induced DNA damage (32, 33, 52). The findings that MUC1-C is necessary for PRC1 and PRC2 function in the DDR thus provide a mechanistic explanation for the effects of targeting MUC1-C on the induction of DSBs and the synergy observed with CDDP treatment.
Of interest for the present work were the previous observations that PRC1 and PRC2 are recruited by PARP1 to sites of DNA lesions (32). PARP1 plays multiple roles in the DDR, including chromatin remodeling and transcriptional regulation (12, 20). PARP1 thus facilitates chromatin relaxation and inhibition of transcription required for the faithful repair of DSBs (12, 20). Our findings that MUC1-C contributes to histone modifications in the DDR invoked the possibility that MUC1-C might also play a role in the regulation of PARP1 activity. MUC1-C interactions with BMI1 and EZH2 and the PARP1-mediated recruitment of these PRC proteins to sites of DNA damage (32) led to the demonstration that MUC1-C also forms a complex with PARP1. Moreover, we found that this association is increased by DNA damage and is not dependent on PARP1 activity. The formation of MUC1-C/PARP1 complexes could be mediated by EZH2, the activity of which is inhibited by PARP1-directed PARylation (30, 53). By contrast, MUC1-C activates EZH2 (29), indicating that MUC1-C and PARP1 may play counteracting roles in the regulation of H3K27me3 levels. In addition to this line of thinking, we found that MUC1-C is of importance for the regulation of PARP1 activation. Targeting MUC1-C genetically and pharmacologically decreased PARP1 activity. One potential explanation for these findings is that MUC1-C promotes the recruitment of BMI1 and EZH2, which are necessary for chromatin relaxation, and their absence decreases PARP1 activity. An alternative, but not mutually exclusive role, is that MUC1-C can directly function in regulating PARP1 activation. As reported for BMI1 and EZH2, PARP1 also recruits the nucleosome remodeling and deacetylation (NuRD) complex to sites of DNA damage (32), invoking the possibility that MUC1-C may similarly contribute to regulation of NuRD components in the DDR. Further studies will thus be needed to define these potentially indirect and/or direct effects of MUC1-C on PARP1 activity; however, the finding that targeting MUC1-C suppresses PARP1 could have additional implications for the clinical development of PARP inhibitors.
A role for MUC1-C in the DDR extends its involvement in driving EMT, the CSC state, anticancer drug resistance, and other hallmarks of the cancer cell (3, 4). Our findings that targeting MUC1-C induces DSBs and is synergistic with CDDP uncover a potential clinical opportunity for designing combinations with genotoxic anticancer agents (4). Previously, we showed that localization of MUC1-C to the mitochondrial outer membrane confers resistance to CDDP and etoposide by blocking the apoptotic response to DNA damage (7). The present results extend those observations by demonstrating that MUC1-C also plays a role in promoting DNA repair and, accordingly, could function in integrating the DDR with induction of apoptosis. Our findings further underscore the potential for targeting MUC1-C in combination with PARP1 inhibitors, which have been approved for the treatment of BRCA-mutated breast and ovarian cancer (16). Functional BRCA1/2 proteins are critical for repair of DSBs (54). However, PARP1 inhibition can be associated with the survival of BRCA reversion mutations (18), emphasizing the need for additional strategies to enhance the effectiveness of olaparib and other PARP inhibitors. Given these clinical challenges, our results indicate that targeting MUC1-C is synergistic with olaparib in settings of both mutant and wild-type BRCA. In this potential therapeutic capacity, the MUC1-C inhibitor GO-203 has been evaluated in a phase I trial as monotherapy for patients with cancer and has demonstrated an acceptable safety profile and evidence of clinical activity. The finding that GO-203 has a short circulating half-life in patients necessitated the reformulation of GO-203 in nanoparticles (GO-203/NPs) for less frequent delivery and more prolonged drug exposure (21). The present findings that GO-203 is effective in combination with olaparib support the premise that targeting MUC1-C could enhance the activity of PARP inhibitors against TNBCs with mutant and wild-type BRCA, consistent in part with a role for MUC1-C in attenuating the formation of DSBs and blocking death of cancer cells in the DDR (7–9). Our findings also provide the experimental basis for further preclinical evaluation of this combination in terms of dosing, number of cycles and effects on survival in additional TNBC, ovarian, and other tumor models.
Disclosure of Potential Conflicts of Interest
D. Kufe is a member of Board of Directors at Genus Oncology, Victa BioTherapeutics, and Nanogen Therapeutics; has ownership interest (including stock, patents, etc.) in Hillstream BioPharma, Nanogen Therapeutics, Victa BioTherapeutics, and Reata; and is a consultant/advisory board member of Reata Pharmaceuticals, CanBas, Genus Oncology, and Victa BioTherapeutics. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions
Conception and design: M. Yamamoto, T. Hata, T. Maeda, M. Miyo, M. Hiraki, K. Hinohara, D. Kufe
Development of methodology: M. Yamamoto, T. Hata, T. Maeda, M. Miyo, M. Hiraki, Y. Suzuki
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): M. Yamamoto, C. Jin, T. Hata, Y. Yasumizu, Y. Zhang, D. Hong, T. Maeda, M. Miyo, H. Rajabi
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): M. Yamamoto, C. Jin, T. Hata, T. Maeda, M. Miyo
Writing, review, and/or revision of the manuscript: M. Yamamoto, T. Hata, D. Hong, M. Miyo, D. Kufe
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): M. Miyo, Y. Suzuki, H. Rajabi
Study supervision: M. Miyo, D. Kufe
Acknowledgments
This work was supported by Grants from the NCI of the NIH under award numbers CA97098, CA166480, CA216553, and CA233084.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.