


Abstract
Eribulin (E7389), a mechanistically unique microtubule inhibitor in phase III clinical trials for cancer, exhibits superior efficacy in vivo relative to the more potent compound ER-076349, a fact not explained by different pharmacokinetic properties. A cell-based pharmacodynamic explanation was suggested by observations that mitotic blockade induced by eribulin, but not ER-076349, is irreversible as measured by a flow cytometric mitotic block reversibility assay employing full dose/response treatment. Cell viability 5 days after drug washout established relationships between mitotic block reversibility and long-term cell survival. Similar results occurred in U937, Jurkat, HL-60, and HeLa cells, ruling out cell type–specific effects. Studies with other tubulin agents suggest that mitotic block reversibility is a quantifiable, compound-specific characteristic of antimitotic agents in general. Bcl-2 phosphorylation patterns parallel eribulin and ER-076349 mitotic block reversibility patterns, suggesting persistent Bcl-2 phosphorylation contributes to long-term cell-viability loss after eribulin's irreversible blockade. Drug uptake and washout/retention studies show that [3H]eribulin accumulates to lower intracellular levels than [3H]ER-076349, yet is retained longer and at higher levels. Similar findings occurred with irreversible vincristine and reversible vinblastine, pointing to persistent cellular retention as a component of irreversibility. Our results suggest that eribulin's in vivo superiority derives from its ability to induce irreversible mitotic blockade, which appears related to persistent drug retention and sustained Bcl-2 phosphorylation. More broadly, our results suggest that compound-specific reversibility characteristics of antimitotic agents contribute to interactions between cell-based pharmacodynamics and in vivo pharmacokinetics that define antitumor efficacy under intermittent dosing conditions. Cancer Res; 71(2); 496–505. ©2011 AACR.
Introduction
Halichondrin B (HB) is a structurally complex marine natural product (1, 2). Although HB's potent anticancer activity (2–4) led to interest by the U.S. National Cancer Institute in developing it as a novel chemotherapeutic, supply of HB from aquaculture prevented such efforts (5). Fortunately, HB's synthesis in 1992 by Kishi and colleagues (6–8), together with discovery that its activity resides in its macrocyclic C1-C38 moiety (Towle MJ, Kishi Y, Littlefield BA, original unpublished data, Eisai Research Institute, 1992; refs. 5, 7, 9, 10), provided both a renewable material source and allowed simplification and optimization. More than 200 analogues of HB's C1-C38 moiety were made (5), culminating in discovery of 2 promising analogues, C35-hydroxyl ER-076349 (NSC-707390) and C35-amine eribulin [NSC-707389 (previous nomenclature includes ER-086526, E7389); ref. 10; Fig. 1]. Eribulin is in phase III clinical trials for advanced metastatic breast cancer and in phase II trials for other cancer types. Clinically, eribulin is administered using intermittent dosing (typically, days 1 and 8 of a 21-day cycle; see www.clinicaltrials.gov for further information), consistent with preclinical indications that it is maximally efficacious when dosed intermittently (11).
![Figure 1. Structures of halichondrin B, eribulin, and ER-076349. *, Tritiums in [3H]eribulin and [3H]ER-076349.](https://aacr.silverchair-cdn.com/aacr/content_public/journal/cancerres/71/2/10.1158_0008-5472.can-10-1874/2/m_496fig1.jpeg?Expires=1716383564&Signature=1RsK16Y5L5VfofzmIhOgkDT~HeVu1hYBaYeRdyD-xgiGRMzjf6jFg1r3TwT9ggscWbbwmG5eu9wPh44cqJzJl9-9RWPjLEt0bOnucY5gIc5SHqwbm7UTD1iB9gbxPIEBPwv4Fco-KhdUoMd6ZBLChI7pjYlEj56KAeGhvXsjDXX~dvTM6~04SDkgHdt6euLmxnYOX1P2VIbETYzdYH6WeDmPk4cW-DtQR-Zt5AZqFOPHYdInBOaRfHAE8J-md5~0Glz4Q3~utcZtg6gx8S-166edKWrwz3PGgwigoRRQShEYzZ1MNHxFP7ltL0nWNdlWp44fiFQlJrwaYDytmUHklA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structures of halichondrin B, eribulin, and ER-076349. *, Tritiums in [3H]eribulin and [3H]ER-076349.
Structures of halichondrin B, eribulin, and ER-076349. *, Tritiums in [3H]eribulin and [3H]ER-076349.
Eribulin works via apoptosis-inducing, microtubule-targeting antimitotic mechanisms (10, 12) involving inhibition of microtubule dynamics by mechanisms distinct from most other tubulin-targeting agents (13–15). Eribulin's mechanistic novelty is noteworthy when considering the original mechanistic studies on HB, which showed it to have a spectrum of biochemical effects on tubulin that was distinct from other tubulin-targeted drugs (16). Thus, HB's mechanistic novelty has been preserved in eribulin.
While selecting between ER-076349 and eribulin as a final candidate, we noted that although both compounds showed sub-to-low nmol/L growth inhibitory potencies, similar abilities to disrupt mitotic spindles and induce mitotic blockade, similar susceptibilities to P-glycoprotein–mediated efflux, and similar in vivo pharmacokinetic properties (10, 17), eribulin was consistently superior in vivo against human tumor xenografts even though ER-076349 is somewhat more potent (10). Insights into this came from cell cycle analyses following drug treatment and washout, which revealed that mitotic blockade by eribulin was irreversible after drug washout, whereas ER-076349's blockade was reversible (5, 17). Although differences in mitotic block reversibility would be inconsequential under continuous exposure conditions in typical cell growth inhibition assays, we hypothesized that they might have considerable impact on in vivo efficacy under intermittent dosing conditions in preclinical xenograft models.
Here we present a cell-based assay allowing quantification of mitotic block reversibility of antimitotic agents under drug treatment and washout conditions. This assay was designed to be an in vitro cell-based surrogate for effects of intermittent dosing in vivo. Using eribulin and ER-076349, we explored relationships between mitotic block reversibility, long-term cell viability, inactivation of Bcl-2 and cellular uptake, and retention of radiolabeled probes. Our results provide a plausible explanation for the in vivo superiority of eribulin relative to the more potent ER-076349. More generally, our results argue for consideration of cell-based pharmacodynamics alongside in vivo pharmacokinetics when evaluating the efficacy of antimitotic drugs administered under intermittent dosing conditions.
Materials and Methods
Compounds and synthesis
Structures of HB, eribulin and ER-076349 are presented in Fig. 1. Eribulin and ER-076349 were synthesized as described (18). Radiolabeling was via regioselective palladium-catalyzed H-T exchange with tritium gas (Lewis BM et al., in preparation; specific activities: [3H]eribulin, 29 Ci/mmol; [3H]ER-076349, 30 Ci/mmol). In principle, tritiation could have occurred at either C19 or C26 olefins, but precedent for C19 regioselectivity had been established by earlier work on terminal olefins using metathesis (19). [3H]Vinblastine sulfate (11.4 Ci/mmol) and [3H]vincristine sulfate (7.4 Ci/mmol) were from Amersham International. Results of studies using [3H]vinblastine and [3H]vincristine were corrected for approximately 1.5-fold difference in specific activities. Corrections for [3H]eribulin and [3H]ER-076349 were not made because their specific activities differed by approximately only 3%.
Cells and cell culture
Cell lines were obtained from American Type Culture Collection (ATCC) and grown under ATCC-recommended conditions. U-937 histiocytic lymphoma cells, HL-60 acute promyelocytic leukemia cells, Jurkat acute T-cell leukemia cells (clone E6–1), and HeLa S3 cells were obtained on 2/15/1990, 7/29/1992, 4/29/1996, and 1/13/1997, respectively. All lines were expanded on receipt and cryopreserved at lowest possible passages using a 2-tiered system, to allow experiments to be done with cells that were never more than 20 passages from thaw, or more than 30 passages from receipt from ATCC. DNA profile authentication of all 4 lines has been reported by ATCC; additionally, ATCC reports cytogenetic analyses and/or isoenzyme characterization for HeLa S3, HL-60, and Jurkat cells. No further cell line authentication was done beyond what was provided by ATCC.
Cell cycle analysis
Aliquots of cell suspensions taken at t = 0 h and t = 10 h in the mitotic block reversibility assay were analyzed by flow cytometry as previously described (10).
Mitotic block reversibility assay
Schematic representation of the mitotic block reversibility assay is shown in Fig. 2. U-937 cells were plated in 22.5 mL volumes in 75 cm2 flasks at 1 × 105 cell/mL and incubated for 30 to 34 hours to allow resumption of log-phase growth (optimal time for resuming log-phase growth determined empirically). Test compounds were added as 2.5 mL of 10× concentrates yielding 0.1 to 1,000 nmol/L final concentrations (1–10,000 nmol/L used for colchicine, colcemid, nocodazole due to lower intrinsic growth inhibitory potencies) in half-log increments; compound-free plates received 2.5 mL of vehicle control. This time point, designated t = −12 h, marked the beginning of the 12-hour pretreatment period; previous studies had shown that 12-hour exposure of U-937 cells to antimitotic agents provides optimal balance between full collection of cells in G2/M and minimal apoptosis induction (10, 12). At t = 0 h, cells were harvested and washed twice with 50 mL prewarmed, pre-5% CO2 equilibrated, compound-free media using centrifugation at 300 × g for 10 minutes, followed by resuspension in 35 mL of the same media; 10 mL aliquots were immediately taken for cell cycle evaluation, with the remaining 25 mL returned to fresh flasks for 10-hour incubation in compound-free media. At t = 10 h, 10 mL aliquots were taken from flasks for cell cycle evaluation, with immediate replenishment using 10 mL of fresh, prewarmed, compound-free media. Remaining cells were incubated for 5 days, with one 80% media replenishment after 48 hours to prevent nutrient depletion. After 5 days, cell viabilities were determined by standard trypan blue exclusion techniques. The 5-day postwashout recovery time was empirically selected for maximal amplification and overgrowth of remaining living cells, thus providing clear demarcation of long-term cell killing effects following transient exposure to test agents.

The timing of each step of the U-937 mitotic block reversibility assay was empirically determined on the basis of thorough investigation of cell cycle phase lengths with and without cell synchronization. We had previously determined that the most accurate way of measuring mitotic completion was not by quantifying loss of cells from G2/M, but rather by measuring cells in the G1 phase at any given time, for the following reasons. Previous studies with bromodeoxyuridine pulse-labeling established G1 phase length in log-phase U-937 cells to be approximately 8 hours (Supplementary Fig. S1). In addition, mitotic synchrony studies of U-937 cells using the highly reversible mitotic blocker nocodazole indicated that after nocodazole release, progression of cells through mitosis and into the G1 phase was complete by 10 hours. Cells measured in the G1 peak thus represented cells that had completed mitosis during the previous 10 hours (Supplementary Fig. S2). The G1 peak method of measuring mitotic completion was found to be superior to quantifying decreases in the G2/M peak itself; this method was thus selected for quantifying mitotic completion in the mitotic block reversibility assay. On the basis of analysis of 289 mitotic block reversibility assays, a complete mitotic block (CMB) is considered one in which no more than approximately 1% of cells are present in the G1 phase (steady-state proportion of G1 cells in log-phase U-937 populations is approximately 40%–45%; Fig. 4). Mitotic block reversibility ratios were calculated by dividing the minimum drug concentration required to maintain CMB at t = 10 h by the minimum concentration required to initially induce CMB at t = 0 h.
The mitotic block reversibility assay involves numerous handling steps (add compounds, perform washouts, take aliquots, etc.) that have the potential to cause decreases in media temperature and CO2 saturation levels that can slow cell cycle progression through different phases, markedly degrading data quality. In our experience, successful use of this assay depends on starting with log-phase cells, working fast at each step, using prewarmed media for washing, minimizing cap opening times and times outside of incubators, and not opening incubator doors during incubations. Attempts to reduce the assay scale from 75 cm2 flasks to 25 cm2 flasks consistently led to degradation of data quality, an observation we attribute to accelerated loss of temperature stability and CO2 equilibrium with smaller flasks and media volumes.
Bcl-2 phosphorylation
Cell aliquots were obtained at t = 0 h and t = 10 h during the mitotic block reversibility assay. Lysates were prepared in 50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 0.5% (v/v) NP40, 1 mmol/L dithiothreitol, 1 mmol/L sodium vanadate, 50 mmol/L sodium fluoride, 40 mmol/L β-glycerol phosphate, 1 mmol/L phenylmethylsulfonyl fluoride and protease inhibitors (diluted 1:500; Protease Inhibitor Cocktail Set III, Calbiochem). Proteins were analyzed using 12.5% SDS-PAGE gels followed by immunoblotting with anti–Bcl-2 monoclonal antibody (Dako #M0887) and horseradish peroxidase-conjugated sheep antimouse IgG as secondary antibody (Amersham #931) to detect Bcl-2 as a 26-kDa band. Phosphorylated Bcl-2 tracks slightly higher (slower) than the unphosphorylated species.
Cellular uptake and washout/retention studies
Evaluation of [3H]eribulin, [3H]ER-076349, [3H]vincristine, and [3H]vinblastine uptake and washout/retention in U-937 cells was done as follows. Tritiated compounds were added to sterile 1.5 mL screw cap microcentrifuge tubes and air dried to remove solvent. After compound resuspension in 100 μL cell culture medium, 22.2 × 106 cells were added in 0.9 mL complete culture medium (including fetal bovine serum, glutamine, and antibiotics). Incubations were at 37°C with frequent vortexing. Compound uptake as a function of drug concentration was determined after 60 minutes, the empirically determined minimum time needed to reach maximal radioactivity uptake. Cell-associated radioactivity was determined as follows. Triplicate 25-μL samples of cells were removed and layered on top of 300 μL ice-cold 20% sucrose in 400 μL Sarstedt tubes, followed by centrifugation at 8,500 × g for 1 minute to separate cells from labeled media and to wash cells during centrifugal transit through the sucrose. Tubes with pelleted cells were immediately frozen in a dry ice/ethanol bath. Bottom portions of the still-frozen tubes containing pellets were immediately cut off directly into scintillation vials, followed by addition of scintillation fluid and radioactivity counting (Beckman LS 6000 counter). Because of unexpectedly large differences in cellular uptake between the paired drugs (Fig. 6), different preloading concentrations were used for washout/retention studies to start with equal intracellular drug levels (dashed lines, Fig. 6A and B). Thus, preloading concentrations for washout/retention studies used 800 nmol/L [3H]eribulin versus 100 nmol/L [3H]ER-076349, and 600 nmol/L [3H]vincristine versus 190 nmol/L [3H]vinblastine.
Compound retention after 60-minute preloading was measured after rapid 1:50 dilution of labeled cells into prewarmed, compound-free complete media followed by incubation at 37°C with agitation. Five replicate 75-μL samples were removed, layered on and centrifuged through sucrose as above, and processed for counting immediately after dilution (0 time) and at intervals over a 3-hour period. Percent residual cell-associated radioactivity was determined by dividing mean counts at each time point by mean counts seen at 0 time. Control studies indicated there was no measurable loss of cell-associated radioactivity during the very brief time required for dilution and initial sampling.
Results
Mitotic block reversibility of ER-076349 and eribulin
Figure 3 presents cell cycle profiles of eribulin and ER-076349 in a representative mitotic block reversibility assay, with minimum concentrations needed to induce CMB (0 hours) or sustain CMB after washout (10 hours) being circled. Eribulin and ER-076349 showed significant differences in their abilities to induce mitotic blockade that remained intact 10 hours postwashout. Importantly, such differences are only apparent when mitotic block reversibility is measured with full dose/response titrations, because sufficiently high concentrations of both compounds can induce irreversible mitotic blockade (≥30 nmol/L ER-076349, ≥10 nmol/L eribulin). CMBs at 0 hour were initially induced by 3 nmol/L ER-076349 and 10 nmol/L eribulin, consistent with ER-076349's greater cell growth inhibitory potency (10). For ER-076349, 10-fold higher concentration (30 nmol/L) was required to sustain CMB 10 hours postwashout, indicating that ER-076349 induces moderately reversible mitotic blockade (reversibility ratio: 30 nmol/L/3 nmol/L = 10). In contrast, CMB initially induced by 10 nmol/L eribulin remained intact 10 hours postwashout, indicating that eribulin induces irreversible mitotic blockade (reversibility ratio: 10 nmol/L/10 nmol/L = 1).

U-937 cell cycle histograms for ER-076349 and eribulin in mitotic block reversibility assay. Circles, minimum drug concentrations inducing CMB at 0 hour or maintaining CMB at 10 hours.
U-937 cell cycle histograms for ER-076349 and eribulin in mitotic block reversibility assay. Circles, minimum drug concentrations inducing CMB at 0 hour or maintaining CMB at 10 hours.
Figure 4 shows average mitotic completion and 5-day cell-viability data for ER-076349 and eribulin from multiple assays. On average, CMB was initially induced by 3 nmol/L ER-076349, yet 30 nmol/L was required to maintain postwashout CMB. In contrast, 10 nmol/L eribulin was sufficient to both initially induce CMB and maintain CMB after washout. Thus, averaged data from many experiments establish ER-076349 as a moderately reversible antimitotic agent (reversibility ratio: 35 nmol/L/3 nmol/L = 12), whereas eribulin is an irreversible antimitotic agent (reversibility ratio: 11 nmol/L/11 nmol/L = 1). Importantly, 5-day cell viabilities tracked not with initial 0 hour CMB, but rather with sustained postwashout 10-hour CMB. Thus, for both agents, concentrations that sustained postwashout CMB were identical to those that led to complete viability loss 5 days later.
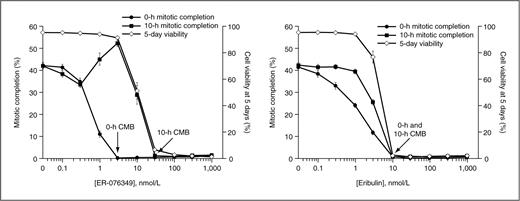
Dose/response of mitotic completion and long-term U-937 cell viability after transient exposure to ER-076349 (left) or eribulin (right) in mitotic block reversibility assay. Results represent means ± SEM from 13 (ER-076349) or 14 (eribulin) separate assays. Methods for determining mitotic completion are described in Materials and Methods.
Dose/response of mitotic completion and long-term U-937 cell viability after transient exposure to ER-076349 (left) or eribulin (right) in mitotic block reversibility assay. Results represent means ± SEM from 13 (ER-076349) or 14 (eribulin) separate assays. Methods for determining mitotic completion are described in Materials and Methods.
Interestingly, ER-076349 showed reproducible postwashout (10 hours) rebound of mitotic completion with maximum at 3 nmol/L (Fig. 4, left), exactly corresponding to the lowest concentration that induced CMB at 0 hour. Because ER-076349–induced blockade is reversible at this concentration, the rebound probably represents synchronous progression of cells through mitosis after recovery from reversible blockade. Rebound is not seen with irreversible eribulin (Fig. 4, right). Thus, cells can be synchronized in mitosis within a narrow 10-fold window using ER-076349, but this is not possible with eribulin.
To rule out cell type-specific effects, mitotic block reversibility studies were done in Jurkat acute T cell leukemia, HL-60 promyelocytic leukemia and HeLa cells (Supplementary Fig. S3). Results from all 3 lines showed that blockade by ER-076349 is reversible, whereas eribulin's blockade is irreversible. We conclude that ER-076349's reversibility and eribulin's irreversibility are compound-specific, cell type–independent characteristics.
Mitotic block reversibility of other tubulin-targeted agents
To test the generality of the mitotic block reversibility concept, reversibility assays were done with paclitaxel, vinblastine, vincristine, colchicine, colcemid, and nocodazole. Results are presented in Table 1, along with data from the eribulin and ER-076349 studies of Figure 4. As already noted, eribulin and ER-076349 were irreversible and moderately reversible mitotic blockers (reversibility ratios: 1 and 12, respectively). Paclitaxel was moderately reversible, (reversibility ratio: 14). Interestingly, vinblastine and vincristine showed widely different reversibility behaviors (reversibility ratios: 65 and 1, respectively); vincristine is thus an irreversible mitotic blocker, like eribulin. Similarly, although colchicine was almost completely irreversible (reversibility ratio: 1.7), colcemid was highly reversible (reversibility ratio: >100). Nocodazole was also highly reversible (reversibility ratio: >100), consistent with its common use as a mitotic synchronizing agent. Overall, results of this survey of tubulin-targeted agents suggest that mitotic block reversibility profiles are unique, quantifiable, and compound-specific characteristics of individual compounds. Importantly, even small structural changes in compounds within a given class can lead to profound differences in mitotic block reversibility (e.g., eribulin versus ER-076349, vincristine versus vinblastine, colchicine versus colcemid).
Mitotic block reversibility ratios of tubulin-targeted agents
| Minimum concentration for CMB, nmol/La | ||||
|---|---|---|---|---|
| Compound | nb | 0 h | 10 h | Reversibility ratioc |
| Eribulind | 14 | 11 | 11 | 1 |
| ER-076349d | 13 | 3 | 35 | 12 |
| Paclitaxel | 3 | 17 | 233 | 14 |
| Vinblastine | 4 | 10 | 650 | 65 |
| Vincristine | 2 | 10 | 10 | 1 |
| Colcemid | 2 | 100 | >10,000 | >100 |
| Colchicine | 3 | 100 | 167 | 1.7 |
| Nocodazole | 2 | 100 | >10,000 | >100 |
| Minimum concentration for CMB, nmol/La | ||||
|---|---|---|---|---|
| Compound | nb | 0 h | 10 h | Reversibility ratioc |
| Eribulind | 14 | 11 | 11 | 1 |
| ER-076349d | 13 | 3 | 35 | 12 |
| Paclitaxel | 3 | 17 | 233 | 14 |
| Vinblastine | 4 | 10 | 650 | 65 |
| Vincristine | 2 | 10 | 10 | 1 |
| Colcemid | 2 | 100 | >10,000 | >100 |
| Colchicine | 3 | 100 | 167 | 1.7 |
| Nocodazole | 2 | 100 | >10,000 | >100 |
aMean values from n experiments.
bNumber separate experiments.
cMean CMB10h/mean CMB0h.
dEribulin and ER-076349 data same as in Fig. 4.
Mitotic block reversibility and Bcl-2 phosphorylation
Because previous studies showed close correlations between Bcl-2 phosphorylation and eribulin-induced apoptosis (12), relationships between eribulin and ER-076349 mitotic block reversibility and Bcl-2 phosphorylation were assessed (Fig. 5). Bcl-2 phosphorylation was not detected in untreated U-937 cells at 0 hour or 10 hours. At 0 hour (after 12 hours pretreatment), Bcl-2 phosphorylation was seen with 0.3 nmol/L ER-076349 and 3 nmol/L eribulin and all higher concentrations of both drugs. In parallel with differences in mitotic block reversibility, maintaining Bcl-2 phosphorylation 10 hour postwashout required 10 nmol/L ER-076349 (33-fold higher than 0 hour), whereas 3 nmol/L eribulin both induced phosphorylation at 0 hour and sustained it 10 hours postwashout. Differences in Bcl-2 phosphorylation thus parallel differences in mitotic block reversibility, linking sustained postwashout mitotic blockade to apoptosis induction and, ultimately, to long-term cell-viability loss.

Bcl-2 phosphorylation data from mitotic block reversibility assay of Fig. 3. Arrows, concentrations at which phosphorylated Bcl-2 (Bcl-2-P) first appears.
Bcl-2 phosphorylation data from mitotic block reversibility assay of Fig. 3. Arrows, concentrations at which phosphorylated Bcl-2 (Bcl-2-P) first appears.
Cellular uptake and washout/retention of [3H]eribulin and [3H]ER-076349
To investigate mechanisms underlying reversibility differences between eribulin and ER-076349, drug uptake and washout/retention studies were done in U-937 cells using tritiated compounds. These studies also included tritiated vincristine and vinblastine as comparators to gauge generalizability of findings. To establish preloading concentrations for washout/retention studies, U-937 cells were incubated for 60 minutes (empirically determined minimum time needed for maximal cell-associated radioactivity) with a range of concentrations of [3H]eribulin, [3H]ER-076349, [3H]vincristine, and [3H]vinblastine. Unexpectedly, the irreversible drugs eribulin and vincristine accumulated to much lower steady-state levels than their reversible counterparts ER-076349 and vinblastine (Fig. 6A and B). Thus, to ensure starting with equal intracellular concentrations for washout/retention studies, cells were preloaded using different extracellular concentrations of the paired drugs: 800 nmol/L [3H]eribulin versus 100 nmol/L [3H]ER-076349, and 600 nmol/L [3H]vincristine versus 190 nmol/L [3H]vinblastine (dashed lines, Fig. 6A and B). Compound washout from cells was achieved via rapid 1:50 dilution with subsequent measurement of cell-associated radioactivity by rapid centrifugation through cold sucrose cushions. Only 10% to 20% of irreversible [3H]eribulin was lost by 180 minutes postwashout, whereas reversible [3H]ER-076349 levels dropped to approximately 35% at 60 minutes and approximately 15% by 180 minutes (Fig. 6C). Similar results were seen with the radiolabeled vincas: only minimal losses of irreversible [3H]vincristine were seen at 180 minutes postwashout, whereas approximately 90% of reversible [3H]vinblastine was lost at 180 minutes postwashout (Fig. 6D).
![Figure 6. Cellular uptake (A, B) and postwashout retention (C, D) of [3H]eribulin and [3H]ER-076349 (A, C), and [3H]vincristine and [3H]vinblastine (B, D). A and B, steady-state accumulation after 60 minutes as a function of loading concentration; C and D, postwashout residual radioactivity relative to t = 0 min (after 60 minutes preloading). Preloading concentrations for C and D were adjusted between paired drugs to start with equal intracellular drug levels; see text.](https://aacr.silverchair-cdn.com/aacr/content_public/journal/cancerres/71/2/10.1158_0008-5472.can-10-1874/2/m_496fig6.jpeg?Expires=1716383564&Signature=RGpOsBVeFI4z8TMe~uZIVwY17HW8It--kbxebaJ--P-Lor-gOBS0IQ455ZcvYIDMSH7AL159~131GqdgGbdgisqAmeFTOPB16gduVTbu3bvCNDbWoQehtfNXMFdTJfSYa7~CHER~2sOeFhowyzJwBdwmzNE54ZoEvknSGhR7jxC6YUz77YiiB3ZoaNJsgSmbuiKGMkHgT6uFhLNU-Vy~l-K67fE1enFRQYDjhTrhJ1xxiTgUZEcxBR7UUW5N1tXhWGl1ce5NjP6KJL~UiJJG5ZFInsm3lIhzuJhPEsPPTcHXajgu1NEYkzgSMziWBaKJjHb2dEcut6MRLyN65lzGkw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cellular uptake (A, B) and postwashout retention (C, D) of [3H]eribulin and [3H]ER-076349 (A, C), and [3H]vincristine and [3H]vinblastine (B, D). A and B, steady-state accumulation after 60 minutes as a function of loading concentration; C and D, postwashout residual radioactivity relative to t = 0 min (after 60 minutes preloading). Preloading concentrations for C and D were adjusted between paired drugs to start with equal intracellular drug levels; see text.
Cellular uptake (A, B) and postwashout retention (C, D) of [3H]eribulin and [3H]ER-076349 (A, C), and [3H]vincristine and [3H]vinblastine (B, D). A and B, steady-state accumulation after 60 minutes as a function of loading concentration; C and D, postwashout residual radioactivity relative to t = 0 min (after 60 minutes preloading). Preloading concentrations for C and D were adjusted between paired drugs to start with equal intracellular drug levels; see text.
Discussion
The aim of this study was to investigate whether cell-based pharmacodynamic effects could explain eribulin's in vivo superiority over ER-076349, an analog with greater in vitro potency. Because previous studies of in vivo pharmacokinetics, chemical stability in serum, and sensitivity to P-glycoprotein–mediated efflux had failed to explain eribulin's in vivo superiority (17), we hypothesized that cellular differences in response to fluctuating drug levels in vivo might provide an explanation. Using a quantitative mitotic block reversibility assay as a cell-based surrogate for in vivo drug level fluctuations, we asked whether eribulin and ER-076349 differed in their abilities to maintain mitotic blockade after drug washout. Because prolonged eribulin-induced mitotic blockade leads to apoptosis (12), the relevance of mitotic block reversibility to long-term cell viability was also examined. We also considered relationships between mitotic block reversibility, Bcl-2 phosphorylation, and drug uptake and retention as potential mechanisms contributing to different pharmacodynamic behaviors of eribulin and ER-076349.
Our results indicate that eribulin induces irreversible mitotic blockade after transient drug exposure, leading to long-term loss of cell viability that tracks closely with dose/response characteristics of the sustained mitotic block. For eribulin, there is no concentration below which an initial CMB becomes reversible postwashout (reversibility ratio: 1). In contrast, ER-076349's mitotic blockade is reversible: approximately 12-fold higher concentrations are required to sustain CMB postwashout (reversibility ratio: 12). Thus, within this approximately 1-log concentration window, ER-076349–treated cells recover from CMB after washout, complete mitosis and repopulate long-term viable cultures. With ER-076349, long-term viability loss occurs only at concentrations high enough to sustain postwashout CMB, that is, 12-fold higher than those initially inducing CMB. The relevance of sustained postwashout CMB, as opposed to initial CMB induction, is thus shown by tight dose/response correlations with long-term viability.
Results with other antimitotic agents (paclitaxel, vinblastine, vincristine, colchicine, colcemid, nocodazole) suggest that mitotic block reversibility is a quantifiable, compound-specific property with relevance to antimitotic agents in general. For instance, paclitaxel is moderately reversible (reversibility ratio: 14), whereas vinblastine, colcemid, and nocodazole show even greater reversibility (reversibility ratios: 65, >100, and >100, respectively). In contrast, and similar to eribulin, vincristine is irreversible and colchicine is nearly so (reversibility ratios: 1.0 and 1.7, respectively). It is noteworthy that, within individual compound classes, dramatic differences in reversibility can occur with only small structural changes. For instance, moderately reversible ER-076349 differs from irreversible eribulin in only one substituent (C35-hydroxyl versus C35-amine, respectively). Similarly, one substituent differentiates highly reversible vinblastine from irreversible vincristine (N-methyl versus N-formyl, respectively), and highly reversible colcemid from nearly irreversible colchicine (N-methyl versus N-acetyl, respectively). Further evidence that small structural changes can elicit large reversibility differences comes from previous studies of many HB analogs. In an analysis of more than 30 analogs of HB's macrocyclic pharmacophore, reversibility ratios of 1 to more than 100 were found (5, 20). Indeed, after early observations that highly reversible analogs lacked in vivo activity, quantifying mitotic block reversibility became an integral part of medicinal chemistry optimization strategies that led to eribulin's discovery (5).
Phosphorylation-dependent inactivation of antiapoptotic Bcl-2 following prolonged activation of the mitotic spindle checkpoint has been implicated in apoptosis induced by antimitotic agents (21–24). Previous studies showed that eribulin-induced Bcl-2 phosphorylation tracks closely with apoptosis induction (12). The current studies extend this by showing that the reversibility of Bcl-2 phosphorylation mimics mitotic block reversibility for eribulin and ER-076349, providing a link between mitotic block reversibility differences and different long-term effects on viability. Thus, sustained Bcl-2 phosphorylation following eribulin's irreversible mitotic block maintains inactivation of Bcl-2's antiapoptotic effects, leading to apoptosis and complete loss of long-term viability at 5 days postwashout.
Results from [3H]eribulin and [3H]ER-076349 uptake and washout/retention studies suggest that eribulin's mitotic block irreversibility results from sustained postwashout cellular retention, whereas ER-076349's reversibility results from rapid postwashout drug loss. These results are consistent with Okouneva and colleagues (14), who showed persistent postwashout [3H]eribulin retention compared with rapid postwashout [3H]ER-076349 loss in U-2 OS osteosarcoma cells. Paradoxically, our results and those of Okouneva and colleagues (14) show that [3H]eribulin accumulates to substantially lower steady-state levels compared with [3H]ER-076349, even though the former is more persistently retained compared with the latter. A similar situation occurred with the 2 vinca alkaloids: Although irreversible [3H]vincristine accumulated to lower steady-state levels, it was more persistently retained compared with reversible [3H]vinblastine's higher initial accumulation, but greater postwashout loss. Overall, our results and those of Okouneva and colleagues (14) are consistent with the idea that the irreversible mitotic blockades induced by eribulin and vincristine derive from persistent cellular retention of these drugs.
Although the basis for differences in drug uptake and postwashout retention is unknown, two potential explanations come to mind. First, this may simply reflect differential efflux via P-glycoprotein or related transporters. However, this seems unlikely for several reasons. First, the P-glycoprotein blocking agent verapamil (25) has no effect on uptake and postwashout retention of either [3H]eribulin or [3H]ER-076349 (Supplementary Fig. S4). Second, eribulin and ER-076349 show similar susceptibilities to P-glycoprotein–mediated efflux (17), arguing against P-glycoprotein as the source of the 2 drugs' different behaviors. Third, any transporter with sufficiently high selectivity for eribulin to result in lower accumulation would be expected to drive enhanced postwashout efflux, yet the opposite was found: eribulin was more persistently retained compared with ER-076349. On the basis of these considerations, it seems unlikely that differential transporter selectivity for eribulin and ER-076349 can account for observed differences in mitotic block reversibility, drug uptake, and postwashout retention.
Another possible explanation involves different tubulin binding characteristics. Our studies showed correlations between reversibility, drug uptake, and postwashout retention for halichondrin analogs eribulin and ER-076349 and vinca alkaloids vinblastine and vincristine. With respect to the halichondrins, Smith and colleagues (15) showed that eribulin binds predominantly to a small number of high affinity sites on microtubule plus ends, with earlier work from that group arguing against significant binding to microtubule sides on the basis of eribulin's lack of effect on microtubule shortening (13). Moreover, eribulin binds microtubule ends with approximately 10-fold greater potency than soluble tubulin heterodimers (15), suggesting that the number of eribulin binding sites in cells is limited primarily by the number of microtubule plus ends. Studies by Alday and Correia (26) showed that ER-076349, but not eribulin, enhances tubulin self-association into oligomers by binding at or near interdimer interfaces, suggesting that ER-076349 can associate with existing oligomers, including microtubule sides. This would dramatically increase the number of intracellular binding sites for ER-076349 relative to eribulin, and explain both our findings and those of Okouneva and colleagues (14) that [3H]ER-076349 accumulates in cells to significantly higher levels than [3H]eribulin. If the lower number of binding sites at microtubule ends were of higher affinity than those along the sides (accessible only to ER-076349), this would also explain higher postwashout retention of [3H]eribulin relative to [3H]ER-076349. Thus, although eribulin would bind to lower numbers of intracellular sites than ER-076349, its overall binding affinity would be greater, resulting in slower off-rates and more persistent postwashout retention. Although this is an attractive hypothesis, further comparative studies of eribulin and ER-076349 binding to tubulin and microtubules will be needed to clarify this issue.
Patterns of mitotic block reversibility, drug uptake, and postwashout retention seen with eribulin and ER-076349 were closely mirrored by similar findings with vincristine and vinblastine. Interestingly, vincristine binds with higher overall affinity to tubulin than vinblastine (27), consistent with our finding of greater postwashout retention of [3H]vincristine relative to [3H]vinblastine. Also, although generally less potent against cells, vincristine more effectively inhibits tubulin addition to microtubules in vitro compared with vinblastine (28), a finding reminiscent of eribulin's lower cell-based potency compared with ER-076349 (10). Although the current studies focused mainly on eribulin and ER-076349, with vincristine and vinblastine used as comparators, the similarities of mitotic block reversibility, drug accumulation, retention, and some known aspects of tubulin binding kinetics suggest that such patterns may be generally applicable among structurally related members within individual classes of tubulin binding agents.
In summary, our results point to eribulin's ability to induce irreversible mitotic blockade as a cell-based pharmacodynamic explanation for its superior in vivo efficacy relative to the more potent ER-076349. Eribulin's irreversible mitotic blockade leads to persistent inactivation of Bcl-2, leading to apoptosis and complete long-term loss of cell viability. Eribulin's irreversible mitotic blockade is associated with lower cellular drug uptake yet more persistent postwashout retention compared with ER-076349, although the precise mechanisms behind this remain unclear. Preferential binding of eribulin to small numbers of high affinity sites at microtubule ends may explain its persistent postwashout retention, although further studies are needed to confirm this. Regardless of mechanism, correlations between eribulin's irreversibility and long-term postwashout cell-viability loss provide a satisfying explanation for its in vivo superiority over ER-076349 in preclinical tumor models using intermittent dosing. In ongoing clinical trials, eribulin is administered using intermittent dosing. Its ability to induce irreversible mitotic blockade in cancer cells may thus have important pharmacodynamic implications for its clinical activity as well, in which fluctuations in circulating drug levels associated with intermittent dosing routinely occur.
Note: Eribulin was approved by the U.S. Food and Drug Administration on November 15, 2010, for use in treating patients with metastatic breast cancer who have received at least two prior chemotherapy regimens, including anthracycline- and taxane-based chemotherapies. Eribulin is marketed by Eisai, Inc. under the trade name Halaven™.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Karleen Nicholson of Complete Medical Communications, who provided medical writing support funded by Eisai, Inc.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.