


Abstract
Nickel is a potent environmental pollutant in industrial countries. Because nickel compounds are carcinogenic, exposure to nickel represents a serious hazard to human health. The understanding of how nickel exerts its toxic and carcinogenic effects at a molecular level may be important in risk assessment, as well as in the treatment and prevention of occupational diseases. Previously, using human and rodent cells in vitro, we showed that hypoxia-inducible signaling pathway was activated by carcinogenic nickel compounds. Acute exposure to nickel resulted in the accumulation of hypoxia-inducible transcription factor (HIF)-1, which strongly activated hypoxia-inducible genes, including the recently discovered tumor marker NDRG1 (Cap43). To further identify HIF-1-dependent nickel-inducible genes and to understand the role of the HIF-dependent signaling pathway in nickel-induced transformation, we used the Affymetrix GeneChip to compare the gene expression profiles in wild-type cells or in cells from HIF-1α knockout mouse embryos exposed to nickel chloride. As expected, when we examined 12,000 genes for expression changes, we found that genes coding for glycolytic enzymes and glucose transporters, known to be regulated by HIF-1 transcription factor, were induced by nickel only in HIF-1α-proficient cells. In addition, we found a number of other hypoxia-inducible genes up-regulated by nickel in a HIF-dependent manner including BCL-2-binding protein Nip3, EGLN1, hypoxia-inducible gene 1 (HIG1), and prolyl 4-hydroxylase. Additionally, we found a number of genes induced by nickel in a HIF-independent manner, suggesting that Ni activated other signaling pathways besides HIF-1. Finally, we found that in HIF-1α knockout cells, nickel strongly induced the expression of the whole group of genes that were not expressed in the presence of HIF-1. Because the majority of modulated genes were induced or suppressed by nickel in a HIF-1-dependent manner, we elucidated the role of HIF-1 transcription factor in cell transformation. In HIF-1α-proficient cells, nickel exposure increased soft agar growth, whereas it decreased soft agar growth in HIF-1α-deficient cells. We hypothesize that the induction of HIF-1 transcription factor by nickel may be important during the nickel-induced carcinogenic process.
INTRODUCTION
The utilization and disposal of nickel (Ni)-containing products in modern industry leads to environmental pollution by both soluble and insoluble forms of Ni. Additionally, combustion of fossil fuel contributes significantly to environmental burden, mostly by producing aerosols containing soluble Ni (1). Human exposure to Ni occurs primarily via inhalation and ingestion (2). Cutaneous exposure to Ni causes allergy in the form of contact dermatitis. The chronic exposure to Ni compounds can lead to asthma, inflammation, lung fibrosis, and kidney diseases, but the most serious concerns relate to the carcinogenic activity of Ni (2). Epidemiological studies have clearly implicated Ni compounds as human carcinogens based on a higher incidence of lung and nasal cancer among Ni mining, smelting, and refinery workers (2, 3). In various animal models, Ni compounds induce tumors at virtually any site of administration (2). Additionally, insoluble Ni compounds such as nickel subsulfide efficiently transform rodent and human cells in vitro (2). Based on these observations, the IARC evaluated the carcinogenicity of Ni in 1990 (4). All Ni compounds except for metallic Ni were classified as carcinogenic to humans.
The molecular basis of Ni carcinogenesis has been challenging because carcinogenic Ni compounds were weakly mutagenic in most assay systems, even though they were able to produce oxidative DNA damage and inhibit DNA repair activity (2, 5, 6, 7, 8, 9). The level of oxidative stress induced by Ni in cells is rather weak when compared with other metals; however it depleted glutathione and activated AP1,3 nuclear factor κB, and other oxidatively sensitive transcription factors (10, 11, 12, 13).
Recently, new data related to the activation of hypoxic signaling pathways by Ni have emerged (13, 14, 15). The response to hypoxia is mediated primarily by the HIF-1 transcription factor. In the presence of oxygen, proline 564 in the HIF-1α subunit is hydroxylated by a prolyl hydroxylase (16). The prolyl hydroxylation of HIF-1α allowed VHL binding, leading to the proteosomal destruction of this protein. The prolyl hydroxylase that fulfills this function requires both oxygen and iron. It is likely that Ni substitutes iron in the prolyl hydroxylase and inactivates the enzyme, thus switching a cell’s metabolism to a state that mimics a state of hypoxia. A first testing of this hypothesis has been initiated by cloning Ni-inducible genes (17). One gene, NDRG1 (Cap43), was found to be highly induced by soluble and insoluble Ni compounds in all tested cell lines (17). In addition to its induction by Ni compounds, NDRG1 (Cap43) was also induced by hypoxia in a HIF-1-dependent manner (18). Moreover, we have shown that acute exposure to Ni activates HIF-1, and in Ni-transformed cells, the activity of this transcription factor is significantly elevated (14). Here, using the Affymetrix GeneChip and GeneSpring analysis software, we show that the exposure of cells to Ni triggered the expression of hypoxia-inducible genes involved in glucose transport and glycolysis. Other hypoxia-inducible genes up-regulated by Ni in a HIF-dependent manner were BCL-2-binding protein Nip3, EGLN1, HIG1, prolyl 4-hydroxylase, and p125FAK (19, 20, 21, 22, 23, 24).
The induction of ATM, p53, and the p53-dependent genes GADD45 and cyclin kinase inhibitor p21 in a HIF-1-independent manner suggests that the p53-dependent pathway was another pathway activated by Ni. The response to the appearance of damaged or unfolded proteins by Ni was HIF-1 independent and can be recognized by the induction of both Chop 10 (GADD153) and chaperone HSP70 (25, 26). Thus, both HIF-dependent and HIF-independent pathways were activated by Ni exposure; however, of 114 genes induced >4-fold by Ni in HIF-1α-proficient cells, 85 genes were induced in a HIF-dependent manner, indicating that HIF-1-dependent pathway was a major pathway affected by Ni. The importance of HIF-1 transcription factor in Ni-induced transformation was tested by the ability of HIF-proficient and -deficient cells exposed to Ni to grow in soft agar. Exposure of HIF-proficient but not HIF-deficient cells to Ni resulted in increased anchorage-independent growth. This study is among the first to suggest that hypoxia-like conditions could cause cellular transformation. Therefore, the exploration of the molecular mechanisms of Ni carcinogenesis provides new prospects for understanding how hypoxia possibly selects and promotes cellular changes toward the malignant phenotype.
MATERIALS AND METHODS
Materials.
Nickel chloride was obtained from Alfa Aesar (Ward Hill, MA). Cell culture media, FCS, glutamine, and antibiotics were obtained from Life Technologies, Inc. (Rockville, MD). The most commonly used chemicals were purchased from Sigma (St. Louis, MO). Murine Genome U74A Array and Test3 Array were obtained from Affymetrix (Santa Clara, CA).
Cell Culture and Exposure Conditions.
MEFs and cells with HIF-1α knockout (MEF HIF-1−/−) were obtained from Dr. R. Johnson (University of California San Diego, San Diego, CA) and were described previously (18). All cells were maintained at 37°C as monolayers in DMEM supplemented with 10% fetal bovine serum in a humidified atmosphere containing 5% CO2 at 37°C. Both HIF-1α-proficient and -deficient cells were exposed to 1 mm NiCl2 for 20 h. These exposure conditions resulted in survival of approximately 70% of both cell lines. Cell survival was evaluated using two methods, trypan blue and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. Cell exposure to hypoxia has been described previously (18).
Nickel-induced Transformation and Soft Agar Growth.
MEFs and MEF HIF-1−/− were subjected to 0.25 or 0.5 mm NiCl2 exposure for nine rounds. One round consisted of 2 days of Ni exposure followed by 2 days of recovery. Nine rounds are needed to make changes more permanent so that they would last over 4 weeks of the soft agar growth. After nine rounds of NiCl2 exposure, both cell lines were viable and had approximately the same growth rate. Both exposed and nonexposed cells were plated in soft agar to determine the efficiency of anchorage-independent growth. Cells (5 × 104) were plated in 5 ml of 0.33% agar in DMEM with 10% fetal bovine serum overlaid onto a solid layer of 0.5% agar in DMEM supplemented with 10% serum. The culture was maintained for 4 weeks, and then the colonies were stained with INT solution (Sigma) and counted using digital camera EDAS 290 Kodak (Rochester, NY). For each dose of Ni, plating was performed in three dishes to determine the mean ± SD percentage of cloning efficiency.
RNA Isolation and Northern Blot or GeneChip Hybridization.
Total RNA was isolated from nickel-exposed and nonexposed cells using Midi spin columns (Qiagen, Valencia, CA) and used for Northern blot analysis or to prepare poly(A) mRNA using Oligotex mRNA mini kit (Qiagen). For GeneChip analysis, double-stranded cDNA was synthesized with a SuperScript Choice System cDNA synthesis kit (Invitrogen, Carlsbad, CA) by using an oligo(dT)24 primer with a T7 RNA polymerase promoter site added to its 3′-end. The isolated cDNA was used for in vitro transcription in the presence of biotin-11-CTP and biotin-16-UTP using the T7 High Yield Transcript Labeling System (Enzo Diagnostics, Farmingdale, NY). A total of 25–50 μg of the cRNA product in buffer [40 mm Tris-acetate, (pH 8.1), 100 mm potassium acetate, and 30 mm magnesium acetate] was fragmented at 94°C for 35 min. The prepared probe was used for hybridization as a hybridization mix with BSA and herring sperm DNA (0.1 mg/ml; Sigma) for 16 h at 45°C. The test3 chip served for evaluation of probe quality as directed by the manufacturer (Affymetrix). The quality of probe was estimated based on the ratio of signal intensity of 3′- to 5′-ends of housekeeping genes, and the ratio was found to be between 1.4 and 1.7. The quality of probe and labeling procedures were also evaluated based on signal intensity, which was high across all four test chips. After evaluation of probe quality, aliquots of the cRNA hybridization mixtures (15 μg of cRNA in 200 μl of hybridization mix) were hybridized to the murine GeneChip U74Av2 array. After hybridization, samples were washed, stained, and scanned according to procedures developed by the manufacturer (Affymetrix).
Northern Blot Hybridization Probes.
Probes for selected genes identified using microarray were obtained by PCR. Isolated mRNA was converted into cDNA with a SuperScript Choice System cDNA synthesis kit (Invitrogen). The following primers were used for PCR amplification: (a) Cyp1b1, 5′-gcccctggcgacgatt-3′ (upper) and 5′-aggttgggctggtcactcat-3′ (lower), annealing temperature 59°C; (b) SGK, 5′-gagcataacgggacaacatct-3′ (upper) and 5′-cactctgcgataggaaaaagc-3′ (lower), annealing temperature 60°C; (c) HSP70, 5′-acgggcgcgacctgaacaag-3′ (upper) and 5′-cgctgagcttgcccttgaga-3′ (lower), annealing temperature 54°C; (d) Zac1, 5′-aaccacctccagacccacgat-3′ (upper) and 5′-ggagctgccaaaaccccaata-3′ (lower), annealing temperature 59°C; (e) prolyl 4-hydroxylase, 5′-caagtgggtctccaacaagt-3′ (upper) and 5′-caaacactggctcctcca-3′ (lower), annealing temperature 55°C; and (f) Chop10, 5′-cttgaccctgcgtcccta-3′ (upper) and 5′-ctgctccttctccttcatgc-3′ (lower), annealing temperature 55°C. Thirty cycles were applied in each case with 1 min of denaturing, 1 min of annealing, and 1-min extension. Final extension was 10 min. All probes obtained by PCR were purified by agarose gel. Plasmid containing Nip3 probe was kindly provided by Dr. R. Bruick. The probe was isolated from XhoI/HindIII-digested plasmid.
Analysis of Gene Expression Data.
Scanned output files were visually inspected for hybridization artifacts and then analyzed with Affymetrix Microarray Suite 4.0. Arrays were scaled to an average intensity of 125 and analyzed independently. The murine genome U74Av2 microarray represents approximately 6000 functionally characterized gene sequences from the mouse UniGene database as well as approximately 6000 ESTs. After elimination of genes and ESTs with absent calls from the analysis, the intensity of signal of the remaining 5310 genes and ESTs was normalized based on the signal of four independent actin and GAPDH genes. The value of each housekeeping gene across the four chips was divided by the median value of the same housekeeping gene of four chips. The resulting ratios were then averaged for each chip. This average ratio was used as a normalization factor for the signal value correction of all genes within each chip. Fold changes were determined by dividing the mean intensity of each nickel-exposed condition by the mean intensity of the control cells.
The GeneSpring 4.1 program (Silicon Genetics, Redwood City, CA) was used to filter gene expression level with changes of ≥4-fold. The results were visualized using a Venn diagram, and known genes with the GenBank accession numbers are listed in tables.
RESULTS
GeneChip Expression Data Analysis.
Microarray primary data were examined using Affymetrix Microarray Suite 4.0 and GeneSpring version 4.1 (Silicon Genetics). To eliminate bias generated from the signals without present calls, these signals were replaced by a 2-fold of the background value computed by the Affymetrix Microarray Suite. Each chip contained ≤10 expressed control genes present as nonmammalian single gene “spikes.” Little variation was observed across the chip series with respect to the standardized spikes, providing support for comparison of expression levels across different chips.
A Venn diagram was applied to further investigate both the number of genes that had a 4-fold change in expression after Ni treatment and the overlap between these genes in HIF-1α-proficient and -deficient cells (Fig. 1). One hundred and fourteen genes were up-regulated by NiCl2 exposure in a HIF-1-dependent manner, and 66 genes were down-regulated in a HIF-1-dependent manner. Twenty-nine genes were up-regulated by NiCl2 in a HIF-1-independent manner, and 31 genes were down-regulated in a HIF-1-independent manner. Therefore, the expression of approximately 75% of genes induced by Ni and >50% of genes suppressed by Ni was HIF-1-dependent. In HIF-1α-deficient cells, more genes were up-regulated (484 genes) and down-regulated (287 genes) by NiCl2, indicating that without HIF-1α, cells modify gene expression in an unbalanced fashion to survive Ni exposure. Because HIF-1 plays a major regulatory and coordinating role, it is conceivable that the expression of a larger number of genes may compensate for the loss of HIF-1 transcription factor.
Nickel Induction of Glycolytic Enzymes and Genes Involved in Glucose Transport.
Most of the genes involved in glucose metabolism and glycolysis are induced by hypoxia and are HIF-1 dependent (27). Previously, using HIF-1α-proficient and -deficient fibroblasts, we have found that exposure of cells to Ni induced genes involved in hypoxic stress only in HIF-1α-proficient cells (18). These data suggested that Ni activated the HIF-1-dependent pathway. Here, using a GeneChip analysis and HIF-1α-proficient and -deficient fibroblasts, we further investigated the induction of genes by Ni. Table 1 shows the induction of genes involved in glucose transport and glycolysis in HIF-1α-proficient and HIF-1α-deficient mouse fibroblasts exposed to 1 mm NiCl2 for 20 h. Of 12 genes tested, 10 genes were induced by Ni exposure in HIF-1α-proficient cells, but not in HIF-1α-deficient cells. Glucose-6-phosphate dehydrogenase and hexokinase I were unchanged in Ni-exposed cells (Table 1). It is interesting to note that these data were in agreement with the literature data because glucose-6-phosphate dehydrogenase and hexokinase I gene expression were not affected by hypoxia (28). Our results indicate that hexokinase II was induced >10-fold by Ni exposure in a HIF-1-dependent manner. It was reported previously (28) that hexokinase II is stimulated by hypoxia in a HIF-1-dependent manner. GAPDH is known to be induced by hypoxia or Ni (18, 29, 30). Here, with the GeneChip data, we found that GAPDH was induced 4.5-fold in HIF-1α-proficient cells, but not in HIF-1α-deficient cells (Table 1).
HIF-1-dependent Induction of Other Genes by Nickel.
Of the 114 genes induced >4-fold by Ni in a HIF-1-dependent manner, over 60% of these genes were ESTs. Nip3 was the only known gene induced >20-fold by Ni in HIF-1α-proficient fibroblasts (Table 2). Recently, it was reported that this gene was highly induced by hypoxia (19). Northern blot analysis confirmed induction of Nip3 expression only in HIF-1α-proficient cells by Ni (Fig. 2). EGLN1 and prolyl 4-hydroxylase are members of the iron-dependent dioxygenase family involved in hydroxylation of proline residues (20, 21, 22). Both genes were induced in a HIF-1-dependent manner (Table 2; Fig. 2). Prolyl 4-hydroxylase, which was involved in the hydroxylation of proline in collagen, was induced >10-fold. HIG1 is a novel hypoxia-inducible gene with unknown function (23). This gene was induced by Ni >13-fold in a HIF-1-dependent manner (Table 2). p125FAK is one of the key elements in a signaling pathway that regulates changes in cell shape and motility in response to mitogenic stimuli or stress. The enzyme was shown to be activated by hypoxia (24). Nickel induced its expression >5-fold in a HIF-1-dependent manner. Thus, our results show that many of the genes known to be induced by hypoxia are also highly induced by Ni in a HIF-1-dependent manner (Table 2).
HIF-1-independent Induction of Gene Expression by Nickel.
In addition to the activation of the HIF-1-dependent pathway, we were able to show that Ni also induces gene expression in a HIF-1-independent manner (Table 2). Hypoxic stress causes induction of p53 possibly followed by apoptosis, depending on the strength of the induction signal (31). Accumulation of p53 or activation of its transcriptional activity requires ATM kinase that phosphorylates the p53 protein following stress (32). Exposure to Ni increases expression of ATM in both HIF-1α-proficient and HIF-1α-deficient mouse fibroblasts. Because p53 accumulation is occurring at the protein level, it is not clear whether the increase in ATM expression plays any role in the induced p53 expression. The accumulation of p53 in Ni-exposed cells was reported previously by us (14). GADD45 and p21 were induced by Ni in both HIF-1α-proficient and HIF-1α-deficient mouse fibroblasts, suggesting that they were up-regulated through a HIF-1-independent pathway (Table 2). The induction of GADD45 and p21 by Ni exposure was confirmed at the RNA and protein level in different cell lines including HIF-1α-proficient and HIF-1α-deficient mouse fibroblasts (data not shown). GADD45 and p21 are transcriptionally dependent on p53 and mediate p53-dependent growth arrest (33, 34). Their induction probably represents a safety mechanism that stops proliferation during Ni-imposed toxicity. Other genes including GADD153 (Chop10) and HSP70 were induced in both HIF-1α-proficient and HIF-1α-deficient mouse fibroblasts (Fig. 2). Based on the GeneChip data, HSP70 expression was induced only 2.3-fold in HIF-1α-proficient cells; however, Northern blot revealed significant induction of this gene in both HIF-1α-proficient and HIF-1α-deficient mouse fibroblasts (Fig. 2). Moreover, its induction in HIF-1α-proficient cells was stronger in comparison with HIF-1α-deficient mouse fibroblasts. The gene was not induced by hypoxia, suggesting that this induction was Ni specific. In contrast, Chop10 was induced more strongly by hypoxia than by Ni in both cell lines (Fig. 2). The Chop10 and HSP70 gene products are involved in the unfolded protein response (25, 26). It is conceivable that by binding to proteins, Ni caused an unfolded protein response.
Jun B was induced by Ni in both HIF-1α-proficient and -deficient mouse fibroblasts (Table 2). This protein is a component of the AP1 transcription factor and is usually a heterodimer made up from members of the Jun and Fos protein families. AP1 transcription factor is activated during hypoxic stress in a HIF-1-independent manner, but it cooperates with HIF-1 (35). The induction of Jun B by hypoxia has been described previously (36).
A significant number of genes were induced in HIF-1α-deficient mouse fibroblasts but not in HIF-1α-proficient cells (Fig. 1). Among these genes are NGF-β, SGK, IP10, CD44, heparin binding EGF-like, melanocortin 1 receptor, Grg1, BCL-2-like, and tubulin-binding protein E-Map-115 (Table 2).
IFN-inducible protein 10 (IP-10) was originally discovered as a chemokine that targets both T cells and natural killer cells (37). The elevation of IP-10 expression has been demonstrated in a number of human diseases, including chronic cirrhosis and biliary atresia. Cytokine-responsive gene-2 (Crg-2), the murine orthologue of IP-10, was induced after CCl4 treatment of the hepatocyte-like cell line AML-12. These data suggest that IP-10 might be a stress response gene. Other genes such as NGF-β, SGK, CD44, heparin binding EGF-like, melanocortin 1 receptor, Grg1, BCL-2-like, and tubulin-binding protein E-Map-115 likely represent stress response genes in cells that have lost HIF-1 transcription factor and therefore lost the normal response pathway to hypoxia-like stress (38, 39).
Gene Suppression by Nickel.
A number of genes were significantly suppressed by Ni in a HIF-1-dependent manner, for example, monocyte chemoattractant protein 1 (MCP-1) was suppressed >20-fold (Table 2). MCP-1 was shown to contribute to macrophage infiltration in human ovarian carcinomas (40). MCP-1 production can be stimulated by tumor necrosis factor α. Hypoxia down-regulates tumor necrosis factor α-induced MCP-1 mRNA and protein in ovarian cancer cells (40). The effect was mimicked by cobalt chloride and desferrioxamine, which is consistent with HIF-dependent down-regulation.
Another gene significantly suppressed by Ni in HIF-1α-proficient cells is the tumor suppressor gene Zac1 (Table 2; Fig. 2). It was shown that Zac1 inhibited proliferation of tumor cells in vitro, as evidenced by measuring colony formation, growth rate, and cloning in soft agar (41, 42). In vivo, Zac1 abrogated tumor formation in nude mice. The antiproliferative activity of Zac1 was due to the induction of extensive apoptosis and of G1 arrest, which proceeded independently of the retinoblastoma protein and the regulation of other cell cycle-related proteins such as p21(WAF1/Cip1), p27Kip1, p57Kip2, and p16INK4a. Zac1 is thus the first gene besides p53 that concurrently induces apoptosis and cell cycle arrest. In HIF-1α-deficient cells, Zac1 was not expressed (Fig. 2).
Neuropilin-1 (Npn-1) expression is suppressed >5-fold by Ni in a HIF-1α-dependent manner (Table 2). It is a transmembrane receptor expressed by both endothelial and nonendothelial cells, including tumor cells (43). Npn-1 has been postulated to function as a cofactor in the activation of the biologically relevant VEGF receptor 2 by VEGF165. However, the function of Npn-1 in normal and pathological angiogenesis, its expression pattern in relation to VEGF in tumors such as astrocytomas, and whether it is similarly or differentially regulated compared with VEGF remain unknown. Hypoxia, the main physiological inducer of VEGF expression, decreased Npn-1 expression (43).
Laminins are major basement membrane proteins in vivo. Laminin 5 plays an important role in regulating the behavior and function of epithelial cells in the skin, lung, kidney, gastrointestinal tract, and many other tissues both in vivo and in vitro. It was shown previously that hypoxic conditions decreased production of laminin-5 γ2 in both keratinocytes and astrocytes (44, 45). We found that laminin 5 γ2 was also 5-fold down-regulated by Ni in a HIF-1-dependent manner (Table 2). Thus, suppression of the same gene by hypoxia and Ni suggested the involvement of a common pathway.
The growth arrest-specific gene Gas-1 was shown to be preferentially expressed in quiescent NIH3T3 cells. Its expression inhibits DNA synthesis, suggesting that Gas-1 may be a tumor suppressor gene (46). This gene was strongly suppressed by Ni in both HIF-1α-proficient and HIF-1α-deficient mouse fibroblasts (Table 3). It was shown previously that GAS1-antisense cells had altered morphology and grew to a much higher saturation density than control cell lines with a loss of contact inhibition (46). However, there was no change in requirements for serum or any development of anchorage independence.
Transformation of HIF-1α-proficient and HIF-1α-deficient Fibroblasts by Nickel.
In vitro nickel compounds display potent transforming capability in both human and rodent cell systems (47, 48, 49). After Ni exposure, most rodent cells were found positive for soft agar growth. Here we evaluated the ability of NiCl2 to stimulate soft agar growth in HIF-1α-proficient and HIF-1α-deficient mouse fibroblasts. Both of these cell lines are already transformed and display low but measurable anchorage-independent growth. Exposure to nine rounds of NiCl2 resulted in dose-dependent increase of colony forming ability in HIF-1-proficient cells and in dose-dependent decrease of colony forming ability HIF-1-deficient cells (Fig. 3).
DISCUSSION
Nickel is a modern environmental contaminant that is toxic and carcinogenic. The mechanism by which Ni induces carcinogenesis is still unknown. One possible pathway may involve changes in DNA methylation (50). Whereas epigenetic mechanisms probably play a significant role in the carcinogenicity of Ni, alternative mechanisms are likely to act in concert with these epigenetic effects. Most carcinogens cause cancer by impacting on a variety of different mechanisms. Previously, we have shown that the expression of a number of genes was altered in Ni-exposed cells (17, 18). The changes in gene expression in exposed cells resulted from the activation of a number of transcription factors including ATF-1, p53, and HIF-1 (12, 14, 18, 51). Accumulation of HIF-1 transcription factor, the master regulator of hypoxic response, suggested that the hypoxia-inducible signaling pathway was affected by Ni. Indeed, we noted previously (18) that a set of hypoxia-inducible genes was activated by Ni in HIF-1α-proficient cells, but not in HIF-1α-deficient cells.
In this study, GeneChip technology was used to further identify (a) genes induced by Ni in HIF-1α-proficient cells but not in HIF-1α-deficient cells, (b) genes induced by Ni in both HIF-1α-proficient and -deficient cells, and (c) genes suppressed by Ni in both HIF-1α-proficient and -deficient cells. This technology allowed us to simultaneously analyze the expression of approximately 12,000 genes and ESTs. We confirmed that many genes involved in glucose transport and glycolysis were induced by Ni in a HIF-1-dependent manner. All of these Ni-induced genes are also known to be induced by hypoxia (52). The expression of two genes, glucose-6-phosphate dehydrogenase and hexokinase I, was not changed in Ni-exposed cells. These genes also were not changed by hypoxic exposure (28). Some genes involved in glycolysis were previously shown to be induced by both Ni and hypoxia. For example, the glycolytic enzyme GAPDH was induced by hypoxia and nickel chloride in endothelial cells (18, 29, 30). Thus, the data obtained regarding the induction of genes regulating glucose transport and glycolysis in HIF-1α-proficient but not in HIF-1α-deficient Ni-exposed cells confirmed our previous observations that Ni activates the HIF-1α-dependent signaling pathway.
Hypoxia plays an important role in tumor progression. It selects for cells with enhanced glycolytic activity, causing production of large amounts of lactic acid, one of the most common features of tumor cells (Warburg effect; Ref. 53). It is clear that exposure to Ni activates the hypoxia-inducible pathway and, like hypoxia, facilitates selection of cells with increased glycolytic activity. In addition to the genes involved in glucose metabolism and glycolysis, a number of other hypoxia-inducible genes were found to be elevated by Ni in a HIF-1α-dependent manner in our GeneChip experiment. For example, Nip3 is a proapoptotic gene significantly induced by hypoxia (19). The microarray data showed a >20-fold induction of this gene in Ni-exposed HIF-1α-proficient cells, but not in HIF-1α-deficient cells. Northern blot analyses confirmed the strong induction of this gene by Ni only in HIF-1α-proficient cells.
EGLN1 is a mouse homologue of the Caenorhabditis elegans egl-9 gene suspected of coding for an enzyme that regulates HIF-1α prolyl hydroxylation (20, 21). Both EGLN1 and prolyl 4-hydroxylase are members of the iron-dependent dioxygenase family involved in proline hydroxylation on proteins. Dioxygenases are iron-dependent enzymes that require oxygen for their enzymatic activity and are up-regulated by hypoxic conditions. Because Ni induces HIF-1 transcription factor, it was not surprising to find that these genes were up-regulated by Ni exposure in a HIF-1-dependent manner. Adhesive interactions between cells and the extracellular matrix play a pivotal role in cell morphology, motility, and growth. One of the key enzymes involved in the maintenance of focal adhesion contacts is tyrosine kinase p125FAK. It is known that enzymatic activity of p125FAK was induced by hypoxia (24). We found here that transcription of p125FAK was up-regulated by Ni in a HIF-1-dependent manner. Thus, these data again demonstrate the similarity in the pattern of gene induction by Ni and hypoxia.
Other genes induced by Ni displayed HIF-independent regulation including p21, p53, and GADD45. Because both p21 and GADD45 were regulated by p53 (33, 34), it is therefore likely that the induction of p53 by Ni enhanced expression of these genes. These data are in agreement with our previous finding where activation of p53-dependent transcription in Ni-exposed cells was noted (14). ATM, p21, GADD153, and Jun B showed mixed regulation. Their inductions in HIF-1α-proficient cells were strong; however, some induction in HIF-1α-deficient cells was also observed. This indicates that other regulatory pathways are involved in regulation of these genes.
Nickel induced or suppressed four times more genes in HIF-1α-deficient cells than in HIF-1α-proficient cells. It is conceivable that the loss of the normal pathway to respond to hypoxia-like stress resulted in the involvement of other mechanisms for cell survival. NGF-β, SGK, IP10, CD44, heparin binding EGF-like, melanocortin 1 receptor, Grg1, BCL-2-like, and tubulin-binding protein E-Map-115 were all induced by Ni only in HIF-1α-deficient cells, probably representing a backup survival program.
HIF-1-dependent suppression of gene expression likely represents gene-based metabolic reprogramming leading to a rescue process in stressed cells. These events usually involve a persistent down-regulation of energy demand and supply pathways in metabolism throughout the hypoxic period. Nickel exposure mimics these conditions, and gene suppression by Ni is likely to be a part of a survival process. Suppression of tumor suppressors such as Zac1 or Gas1 by Ni probably plays an important role in Ni-induced cell transformation. Thus, suppression of Zac1 not only inactivates this tumor suppressor but also impairs the transcriptional activity of p53 because Zac1 functions together with p53 as a coactivator of responsive genes in cells expressing wild-type p53 (54).
The changes in gene expression after 20 h of Ni exposure were transient and disappeared after Ni removal; however, it is known that chronic exposure to Ni leads to a selection of cells in which these changes are inherited. For example, chronic exposure to Ni leads to selection of cells with high levels of HIF-1 transcriptional activity (14). To assess the role of HIF-1 transcription factor in Ni-induced transformation, HIF-1α-proficient and -deficient cells were treated with Ni for nine rounds and then plated on soft agar (because anchorage-independent growth is considered an important in vitro test for cell transformation). We found that Ni transformed cells only in the presence of HIF-1; in its absence, cells exposed to Ni decreased their ability to grow in soft agar. Because cells were allowed to recover before plating, it was concluded that the reduced plating efficiency in soft agar was unlikely related to toxicity. Nine rounds of exposure to Ni is a long time period; thus, there was ample opportunity for cells to acquire altered phenotypes. Our results therefore emphasize the importance of HIF-1 in directing an orderly response to environmental stressors, such as hypoxia or Ni, and in its absence the cell responds differently and in a disorganized fashion. As measured by gene expression analyses, there were 4–5 times more genes induced or suppressed in HIF-1α-deficient cells as compared with that in cells having HIF-1α. It was also found that in the absence of HIF-1α, the mouse cells were genetically unstable, and thus, with continued passaging, they changed phenotype. Additionally, because Ni is known to affect reprogramming of gene expression, possibly due to changes in histone acetylation (55), it is conceivable that genetic instability of these cells was amplified after Ni exposure. The obtained data suggest that HIF-1 might be an important driving force for cell transformation under hypoxic conditions. Studies of the induction of cell transformation by hypoxia are currently under way.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Supported by Grants ES00260, ES05512, ES10344, and T32 ES07324 from the National Institute of Environmental Health Sciences; Grant R-827351 from the Environmental Protection Agency; and Grant CA16087 from the National Cancer Institute.
The abbreviations used are: AP1, activator protein 1; HIF, hypoxia-inducible transcription factor; MEF, mouse embryo fibroblast; EST, expressed sequence tag; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; EGF, epidermal growth factor; VEGF, vascular endothelial growth factor.
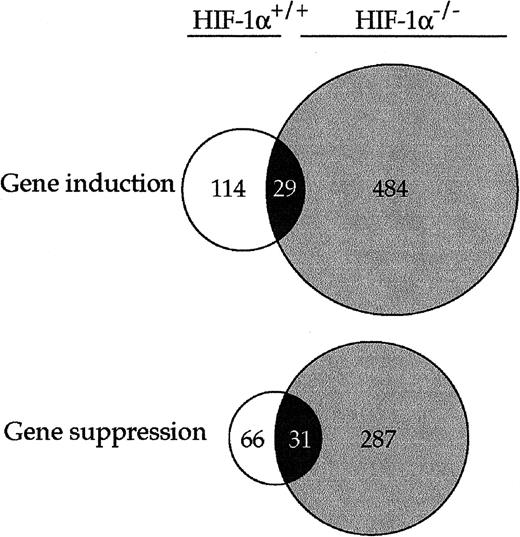
Venn diagram of genes induced or suppressed >4-fold in HIF-proficient and HIF-1-deficient cells. A total of 5285 expressed genes related to two different phenotypes have been filtered using 4-fold change threshold. Open circles represent genes induced or suppressed in HIF-1-proficient cells. Gray-shaded circles represent genes induced or suppressed in HIF-1-deficient cells. Overlapping, black-shaded areas represent genes simultaneously induced or suppressed in both cell lines.
Venn diagram of genes induced or suppressed >4-fold in HIF-proficient and HIF-1-deficient cells. A total of 5285 expressed genes related to two different phenotypes have been filtered using 4-fold change threshold. Open circles represent genes induced or suppressed in HIF-1-proficient cells. Gray-shaded circles represent genes induced or suppressed in HIF-1-deficient cells. Overlapping, black-shaded areas represent genes simultaneously induced or suppressed in both cell lines.

Northern blot analysis of genes differentially expressed in microarray. HIF-proficient and HIF-1-deficient cells were exposed to 0.5 mm NiCl2 or hypoxia (1% O2) for 20 h. Total RNA was isolated and hybridized with the specific probe. Ethidium bromide staining of small ribosomal subunit is shown to assure for equivalent loading.
Northern blot analysis of genes differentially expressed in microarray. HIF-proficient and HIF-1-deficient cells were exposed to 0.5 mm NiCl2 or hypoxia (1% O2) for 20 h. Total RNA was isolated and hybridized with the specific probe. Ethidium bromide staining of small ribosomal subunit is shown to assure for equivalent loading.

Anchorage-independent growth of nickel-exposed cells. HIF-proficient and HIF-1-deficient cells were exposed to NiCl2 as indicated in “Materials and Methods” and plated in soft agar. The histogram shows plating efficiency presented as a percentage of control.
Anchorage-independent growth of nickel-exposed cells. HIF-proficient and HIF-1-deficient cells were exposed to NiCl2 as indicated in “Materials and Methods” and plated in soft agar. The histogram shows plating efficiency presented as a percentage of control.
Changes in the expression of glycolytic enzymes in NiCl2 exposed HIF-1α proficient and -deficient fibroblasts
| Glycolytic enzyme (GenBank accession number) | Fold of increase in HIF-1α-proficient fibroblasts | Fold of increase in HIF-1α-deficient fibroblasts |
|---|---|---|
| Glucose transporter I (M22998) | 8.273 | 0.381 |
| Hexokinase I (J05277) | 1.954 | 1.564 |
| Hexokinase II (Y11666) | 10.42 | 0.459 |
| Glucose-6-phosphate dehydrogenase (Z11911) | 1.09 | 0.56 |
| Glucose phosphate isomerase I (L09104) | 2.233 | 0.723 |
| Phosphofructo kinase B (J03928) | 5.801 | 0.327 |
| Aldolase A (Y00516) | 3.992 | 1.361 |
| GAPDH (M32599) | 4.529 | 0.751 |
| Pyruvate kinase 3 (X97047) | 2.855 | 1.59 |
| Phosphoglycerate kinase I (M15668) | 4.241 | 0.992 |
| Lactate dehydrogenase A (M17516) | 6.443 | 0.805 |
| Triosephosphate isomerase (L31777) | 2.476 | 0.642 |
| Glycolytic enzyme (GenBank accession number) | Fold of increase in HIF-1α-proficient fibroblasts | Fold of increase in HIF-1α-deficient fibroblasts |
|---|---|---|
| Glucose transporter I (M22998) | 8.273 | 0.381 |
| Hexokinase I (J05277) | 1.954 | 1.564 |
| Hexokinase II (Y11666) | 10.42 | 0.459 |
| Glucose-6-phosphate dehydrogenase (Z11911) | 1.09 | 0.56 |
| Glucose phosphate isomerase I (L09104) | 2.233 | 0.723 |
| Phosphofructo kinase B (J03928) | 5.801 | 0.327 |
| Aldolase A (Y00516) | 3.992 | 1.361 |
| GAPDH (M32599) | 4.529 | 0.751 |
| Pyruvate kinase 3 (X97047) | 2.855 | 1.59 |
| Phosphoglycerate kinase I (M15668) | 4.241 | 0.992 |
| Lactate dehydrogenase A (M17516) | 6.443 | 0.805 |
| Triosephosphate isomerase (L31777) | 2.476 | 0.642 |
HIF-dependent and -independent activation of gene expression by NiCl2
| Gene regulation | Gene name (Genbank accession number) | Fold of increase in HIF-1α proficient cells | Fold of increase in HIF-1α deficient cells |
|---|---|---|---|
| HIF dependent | Nip3 (AF041054) | 21.3 | 0.98 |
| HIF dependent | EGLN1 (AI850202) | 14.3 | 0.31 |
| HIF dependent | Hig1 (AV372261) | 13.2 | 0.74 |
| HIF dependent | Prolyl 4-hydroxylase (U16163) | 10.0 | 0.96 |
| HIF dependent | Focal adhesion kinase (M95408) | 5.8 | 0.98 |
| HIF independent | GADD 45 (U00937) | 9.4 | 12.6 |
| HIF independent | P21 (U09507) | 7.8 | 7.8 |
| HIF independent | ATM (U43678) | 6.0 | 2.6 |
| HIF independent | P53 (AB021961) | 2.2 | 2.0 |
| HIF independent | GADD153 (X67083) | 4.3 | 2.8 |
| HIF independent | Jun B (U20735) | 9.6 | 2.4 |
| Induced in HIF-1α-deficient cells | HSP 70 (M12571) | 2.3 | 93.3 |
| Induced in HIF-1α-deficient cells | NGFb (M17298) | 1.8 | 21.9 |
| Induced in HIF-1α-deficient cells | IP-10 (M33266) | 1.1 | 12.1 |
| Induced in HIF-1α-deficient cells | CD44 antigen (X66084) | 1.1 | 10.0 |
| Induced in HIF-1α-deficient cells | Melanocortin 1 receptor (X65635) | 1.1 | 9.0 |
| Induced in HIF-1α-deficient cells | Heparin-binding EGF-like (L07264) | 1.3 | 8.2 |
| Induced in HIF-1α-deficient cells | SGK kinase (AF139368) | 1.8 | 8.0 |
| Induced in HIF-1α-deficient cells | Grg1 (U61362) | 1.3 | 5.0 |
| Induced in HIF-1α-deficient cells | BCL-2-like (L35049) | 1.0 | 4.4 |
| Induced in HIF-1α-deficient cells | E-MAP-115 (Y15197) | 0.46 | 4.1 |
| Gene regulation | Gene name (Genbank accession number) | Fold of increase in HIF-1α proficient cells | Fold of increase in HIF-1α deficient cells |
|---|---|---|---|
| HIF dependent | Nip3 (AF041054) | 21.3 | 0.98 |
| HIF dependent | EGLN1 (AI850202) | 14.3 | 0.31 |
| HIF dependent | Hig1 (AV372261) | 13.2 | 0.74 |
| HIF dependent | Prolyl 4-hydroxylase (U16163) | 10.0 | 0.96 |
| HIF dependent | Focal adhesion kinase (M95408) | 5.8 | 0.98 |
| HIF independent | GADD 45 (U00937) | 9.4 | 12.6 |
| HIF independent | P21 (U09507) | 7.8 | 7.8 |
| HIF independent | ATM (U43678) | 6.0 | 2.6 |
| HIF independent | P53 (AB021961) | 2.2 | 2.0 |
| HIF independent | GADD153 (X67083) | 4.3 | 2.8 |
| HIF independent | Jun B (U20735) | 9.6 | 2.4 |
| Induced in HIF-1α-deficient cells | HSP 70 (M12571) | 2.3 | 93.3 |
| Induced in HIF-1α-deficient cells | NGFb (M17298) | 1.8 | 21.9 |
| Induced in HIF-1α-deficient cells | IP-10 (M33266) | 1.1 | 12.1 |
| Induced in HIF-1α-deficient cells | CD44 antigen (X66084) | 1.1 | 10.0 |
| Induced in HIF-1α-deficient cells | Melanocortin 1 receptor (X65635) | 1.1 | 9.0 |
| Induced in HIF-1α-deficient cells | Heparin-binding EGF-like (L07264) | 1.3 | 8.2 |
| Induced in HIF-1α-deficient cells | SGK kinase (AF139368) | 1.8 | 8.0 |
| Induced in HIF-1α-deficient cells | Grg1 (U61362) | 1.3 | 5.0 |
| Induced in HIF-1α-deficient cells | BCL-2-like (L35049) | 1.0 | 4.4 |
| Induced in HIF-1α-deficient cells | E-MAP-115 (Y15197) | 0.46 | 4.1 |
Suppression of gene expression by NiCl2
| Gene regulation | Gene name (GenBank accession number) | Fold suppression in HIF-1α-proficient cells | Fold suppression in HIF-1α-deficient cells |
|---|---|---|---|
| HIF dependent | MCP-1 (M19681) | 21.2 | 1.3 |
| HIF dependent | Zac 1 (X95503) | 17.5 | 1.1 |
| HIF dependent | Neuropilin (D50086) | 5.6 | 1.1 |
| HIF dependent | Laminin 5 γ2 (U43327) | 5.0 | 1.0 |
| HIF independent | IGF-binding protein 4 (X76066)a | 10.2 | 11.6 |
| HIF independent | Cyp1B1 (X78445) | 21.7 | 30.3 |
| HIF independent | Gas1 (X65128) | 15.8 | 43.4 |
| Gene regulation | Gene name (GenBank accession number) | Fold suppression in HIF-1α-proficient cells | Fold suppression in HIF-1α-deficient cells |
|---|---|---|---|
| HIF dependent | MCP-1 (M19681) | 21.2 | 1.3 |
| HIF dependent | Zac 1 (X95503) | 17.5 | 1.1 |
| HIF dependent | Neuropilin (D50086) | 5.6 | 1.1 |
| HIF dependent | Laminin 5 γ2 (U43327) | 5.0 | 1.0 |
| HIF independent | IGF-binding protein 4 (X76066)a | 10.2 | 11.6 |
| HIF independent | Cyp1B1 (X78445) | 21.7 | 30.3 |
| HIF independent | Gas1 (X65128) | 15.8 | 43.4 |
IGF, insulin-like growth factor.
Acknowledgments
We acknowledge the excellent technical skills of T. Kluz. We are grateful to Dr. R. Bruick for providing us with the Nip3 probe.