

Abstract
We report that PTEN-deficient prostate cancer cells use macropinocytosis to survive and proliferate under nutrient stress. PTEN loss increased macropinocytosis only in the context of AMPK activation, revealing a general requirement for AMPK in macropinocytosis and a novel mechanism by which AMPK promotes survival under stress. In prostate cancer cells, albumin uptake did not require macropinocytosis, but necrotic cell debris proved a specific macropinocytic cargo. Isotopic labeling confirmed that macropinocytosed necrotic cell proteins fueled new protein synthesis in prostate cancer cells. Supplementation with necrotic debris, but not albumin, also maintained lipid stores, suggesting that macropinocytosis can supply nutrients other than amino acids. Nontransformed prostatic epithelial cells were not macropinocytic, but patient-derived prostate cancer organoids and xenografts and autochthonous prostate tumors all exhibited constitutive macropinocytosis, and blocking macropinocytosis limited prostate tumor growth. Macropinocytosis of extracellular material by prostate cancer cells is a previously unappreciated tumor–microenvironment interaction that could be targeted therapeutically.
Significance: As PTEN-deficient prostate cancer cells proliferate in low-nutrient environments by scavenging necrotic debris and extracellular protein via macropinocytosis, blocking macropinocytosis by inhibiting AMPK, RAC1, or PI3K may have therapeutic value, particularly in necrotic tumors and in combination with therapies that cause nutrient stress. Cancer Discov; 8(7); 866–83. ©2018 AACR.
See related commentary by Commisso and Debnath, p. 800.
This article is highlighted in the In This Issue feature, p. 781
Introduction
Cancer cells upregulate nutrient acquisition pathways to fuel oncogene-driven anabolism and proliferation (1). However, as tumors grow, their abnormal vasculature leads to the development of areas of extracellular nutrient limitation. Nutrient import pathways become substrate limited and fail to meet nutrient demand, leading to tumor necrosis (2). Macropinocytosis, a process by which cells nonselectively engulf extracellular material via plasma membrane ruffling (3–5), allows cancer cells with activating mutations in RAS to use extracellular proteins such as albumin as fuel when amino acids are limiting (6–9). Downstream of RAS, PI(3,4,5)P3 (PIP3) and RAC1-GTP are both necessary for macropinosome formation. RAC1 activation induces the actin remodeling and membrane ruffling necessary to form macropinosomes by activating PAK kinases (10, 11). PIP3 produced by type I PI3Ks is required for macropinosome closure; in some cell types, PIP3 is also required for membrane ruffling (12, 13). The lipid phosphatase PTEN opposes PI3K pathway signaling by converting PIP3 to PI(4,5)P2 (14). PTEN is the most frequently deleted tumor suppressor gene in prostate cancer (15, 16); monoallelic PTEN deletion occurs in up to 60% of localized prostate cancers, and complete loss of PTEN is commonly associated with increased risk of metastasis and the development of lethal, castration-resistant disease (16, 17). Consistent with the central role of PIP3 in macropinocytosis, we report that PTEN-deficient prostate cancer cells use macropinocytosis to support anabolism and survival in nutrient-limiting environments. Interestingly, PTEN loss was not sufficient to trigger macropinocytosis in all contexts, revealing a previously unappreciated requirement for AMPK activation to support RAC1 activation and macropinosome formation. Robust macropinocytosis-independent albumin uptake in prostate cancer cells also led to the discovery that necrotic cell debris is a specific macropinocytic cargo, suggesting that macropinocytosis can supply many nutrients, not just amino acids. Taken together, these studies provide critical insights into how nutrient-responsive signaling pathways coordinate the adaptive response to nutrient limitation and suggest a novel mechanism by which the microenvironment affects prostate tumor growth.
Results
PTEN Loss Promotes Macropinocytosis in Fibroblasts under Nutrient-Limiting Conditions
Oncogenic mutations constitutively drive anabolism and limit adaptive metabolic changes under nutrient stress (18). Nevertheless, tumor cells with activating mutations in RAS are paradoxically resistant to amino acid deprivation because they can degrade macropinocytosed proteins in the lysosome to produce amino acids (6–8). PI3K pathway activation is essential for macropinocytosis downstream of RAS (4, 5, 19). To assess whether activating the PI3K pathway would be sufficient to drive macropinocytosis and confer resistance to nutrient stress, uptake of the macropinocytic cargos 70 kD dextran and bovine serum albumin (BSA) was measured in PTEN-null and wild-type murine embryonic fibroblasts (PTEN KO and WT MEFs, respectively) in the presence or absence of the NHE inhibitor 5-(N-ethyl-N-isopropyl) amiloride (EIPA). The dextran or BSA index (percentage of cell area that contains dextran or BSA) was calculated using ImageJ and established protocols (Supplemental Methods; Supplementary Fig. S1A; ref. 20). EIPA inhibits RAC1 activation indirectly by reducing the submembranous pH (21). Although EIPA has pleiotropic effects on cells (22), it is selective for macropinocytosis among endocytic pathways and does not inhibit the clathrin-mediated endocytosis of the EGFR or the transferrin receptor (23, 24). Neither PTEN WT nor PTEN KO MEFs took up dextran or BSA in complete medium (Fig. 1A–C). However, incubation in medium containing only 1% the amount of glucose and amino acids present in standard DMEM (1% AA/gluc) dramatically enhanced both dextran and BSA uptake selectively in PTEN KO MEFs. This increased uptake was sensitive to EIPA, the RAC inhibitor EHT1864, the PAK inhibitor FRAX597, and to dominant-negative RAC1T17N expression (Fig. 1A, C, D; Supplementary Fig. S1B), consistent with internalization via macropinocytosis. Chemical inhibitors of PI3K and PTEN confirmed that PTEN's catalytic activity suppresses macropinocytosis. Inhibiting PTEN with bpV(pic) was sufficient to stimulate macropinocytosis in PTEN WT MEFs in low-nutrient medium (Fig. 1E) but not complete medium. Conversely, inhibiting class I PI3K with a combination of the α or β isoform-selective inhibitors BYL719 and AZD8186 prevented macropinocytosis in PTEN KO MEFs in 1% AA/gluc (Fig. 1F). Together, these results suggest that PI3K pathway activation by PTEN loss is sufficient to promote macropinosome formation selectively under nutrient-limiting conditions.
When macropinocytic cells are subjected to amino acid limitation, albumin supplementation (2%–5%) stimulates proliferation (6–8). A caveat to this approach is that albumin also enters cells through receptor-mediated endocytosis (RME), and supplementation with BSA promotes survival and proliferation even in nonmacropinocytic cells, albeit to a lesser degree (7). Similar to published data from control LSL and KRASG12D MEFs (7), supplementation with 2% BSA increased proliferation of both PTEN WT and KO MEFs in 1% AA/gluc medium, although macropinocytic PTEN KO MEFs proliferated more (Fig. 1G). The value of macropinocytosis was much more apparent in unsupplemented 1% AA/gluc medium, as macropinocytic PTEN KO MEFs were able to proliferate whereas nonmacropinocytic PTEN WT MEFs died. Albumin is the principal protein in fetal calf serum (Supplementary Fig. S2A). Because BSA uptake by macropinocytosis was much more efficient than uptake by RME (Fig. 1A and C), the 0.3% albumin contributed by the serum in the 1% AA/gluc medium was likely sufficient to support survival only in macropinocytic PTEN KO MEFs. Macropinocytic KRASG12D-expressing MEFs, but not matched nonmacropinocytic LSL MEFs, also survived in unsupplemented 1% AA/gluc medium (Supplementary Fig. S2B). In both KRASG12D MEFs and PTEN KO MEFs, this survival advantage was fully reversed by EIPA (Fig. 1H; Supplementary Fig. S2B and S2C). Importantly, EIPA was minimally and equally toxic to macropinocytic and nonmacropinocytic MEFs in complete medium (Fig. 1H; Supplementary Fig. S2B). Similar to EIPA, EHT1864 (allosteric RAC inhibitor) and FRAX597 (PAK inhibitor) were selectively toxic to PTEN KO MEFs relying on macropinocytosis for nutrients (Supplementary Fig. S2D and S2E). Furthermore, reconstitution with PTEN blocked macropinocytosis and eliminated the survival advantage of PTEN KO MEFs in low-nutrient medium (Fig. 1I; Supplementary Fig. S2F). Taken together, these results demonstrate that macropinocytosis confers a survival and proliferative advantage on PTEN KO MEFs in low-nutrient medium.
AMPK Activation Is Necessary for Macropinocytosis
Unexpectedly, PTEN KO MEFs that proliferated in 1% AA/gluc medium (Figs. 1G and 2A) died when deprived of only amino acids (Fig. 2A; Supplementary Fig. S3A). This result suggested that glucose withdrawal stimulated growth. Cells sense and respond to glucose depletion by activating AMPK (25). Strikingly, the allosteric AMPK activator A769662 stimulated robust proliferation in 1% AA medium in PTEN KO MEFs but not in PTEN WT MEFs (Fig. 2A). AMPK promotes the macropinocytosis-dependent entry of Ebola and vaccinia viruses (26, 27). Either glucose depletion or A769662 was sufficient to stimulate dextran uptake in PTEN KO but not WT MEFs (Fig. 2B; Supplementary Fig. S3B). In contrast, amino acid depletion failed to trigger macropinocytosis in PTEN KO MEFs (Fig. 2B). These results suggest that PTEN loss is not sufficient to drive macropinocytosis; AMPK activation is also necessary. Consistent with this model, PTEN deletion from MEFs lacking both AMPK catalytic subunit isoforms (AMPK DKO; ref. 28) failed to trigger macropinocytosis in 1% AA/gluc medium (Fig. 2C; Supplementary Fig. S3C and S3D). The expression of a dominant-negative AMPK (DN-AMPK) mutant or treatment with the AMPK inhibitor compound C also blocked macropinocytosis in PTEN KO MEFs in 1% AA/gluc (Supplementary Fig. S3E and S3F). Although glucose deprivation or A769662 was sufficient to stimulate dextran uptake in PTEN KO MEFs in the presence of normal amino acid levels, colocalization of dextran and Lysotracker Red was reduced relative to 1% AA/gluc (Fig. 2B). This result is consistent with previous reports that mTORC1 inactivation is necessary for efficient macropinosome–lysosome fusion in MEFs (7, 29). In keeping with its role in macropinocytosis, AMPK was necessary for PTEN-null cells to proliferate in 1% AA/gluc medium (Fig. 2D). Taken together, these results demonstrate that AMPK activation is necessary for PTEN-deficient MEFs to form macropinosomes and proliferate under nutrient-limiting conditions.
AMPK activation drives autophagy by activating ULK1 and inactivating mTORC1 (30–32). Autophagy is often necessary for maximal tumor growth (33). To dissect the relative contribution of autophagy and macropinocytosis to AMPK-driven cell proliferation in low nutrients (Fig. 2A), dominant-negative PTENC124S was introduced into MEFs that were either capable of autophagy but deficient in macropinocytosis due to PAK1 deletion or capable of macropinocytosis but deficient in autophagy due to ATG5 deletion. PTENC124S expression in either PAK1 WT or ATG5 WT MEFs led to macropinocytosis upon AMPK activation with A769662 similar to results in PTEN-null MEFs (Fig. 2E and F; Supplementary Fig. S4A–S4D). Deletion of PAK1, but not ATG5, eliminated macropinocytosis. Conversely, deletion of ATG5, but not PAK1, blocked autophagy (Supplementary Fig. S4E–S4G). Consistent with their effect on macropinocytosis (Fig. 2E), PTENC124S expression combined with A769662 drove proliferation in PAK1 WT, but not PAK1 KO, MEFs in amino acid–deficient medium (Fig. 2G). Compensatory upregulation of autophagy was not observed when macropinocytosis was blocked by PAK1 deletion; however, autophagy was slightly elevated basally in PAK1 KO MEFs relative to PAK1 WT controls (Supplementary Fig. S4E). Intriguingly, A769662 stimulated proliferation in both ATG5 WT and KO MEFs expressing PTENC124S in low amino acids (Fig. 2H). Autophagy-deficient PTENC124S ATG5 KO MEFs may have proliferated less than autophagy-competent PTENC124S ATG5 WT MEFs, but autophagy was not necessary for proliferation in amino acid–deficient medium. Taken together, these studies demonstrate that AMPK drives proliferation in nutrient-stressed cells by inducing macropinocytosis. Although autophagy may promote proliferation by sparing macropinocytosis-derived nutrients for anabolic processes, cell-autonomous autophagy is not sufficient to support cell division.
How AMPK stimulates macropinosome formation was next investigated. AMPK activates RAC1 in certain contexts (34), and RAC1 activation is required for membrane ruffling during macropinocytosis (4, 5, 19). We measured RAC1-GTP levels and localization using a dual-chain RAC1 Förster resonance energy transfer (FRET) biosensor and fluorescence lifetime imaging microscopy (FLIM; ref. 35). When the biosensor (PAK1 effector domain–YPET fusion) binds CyPet-RAC1-GTP, the resulting FRET quenches the fluorescence lifetime of the donor (CyPET). Detecting FRET by monitoring donor lifetime rather than following the ratio of acceptor:donor fluorescence intensity has the key advantage that measurements are independent of protein concentration, whereas the phasor approach to fluorescence lifetime data analysis can provide a global view of the RAC1 activation state in an image by transforming the histogram of time delays in each pixel into a phasor (Supplementary Fig. S5A). When the AMPK activator A769662 was added to PTEN KO MEFs in complete medium, RAC1-GTP levels increased, particularly in the cell periphery (Fig. 2I; Supplementary Fig. S5B and S5C). A769662 also stimulated robust AMPK and RAC1 activation and membrane ruffling in PTEN WT MEFs, indicating that PTEN deficiency was not required for RAC1 activation. Macropinosome closure leads to RAC1 inactivation (36). Thus, productive macropinosome formation in PTEN KO MEFs (Fig. 1A) may lead to reduced total RAC1-GTP levels relative to PTEN WT MEFs. Confirming the specificity of the assay, RAC1-GTP levels did not increase in the absence of A769662, and adding the RAC inhibitor EHT1864 60 minutes after A769662 restored donor lifetime to basal levels (Supplementary Fig. S5D and S5E). Using standard ratiometric techniques to monitor the localization and dynamics of RAC1 activation in real time, dynamic waves of RAC1-GTP were seen in the periphery of A769662-stimulated PTEN WT and KO cells; consistent with dextran uptake results, circular macropinosome-like structures bounded by activated RAC1 were observed only in PTEN KO MEFs (Fig. 2J; Supplementary Videos S1–S2). Thus, AMPK activation promotes macropinocytosis by increasing RAC1 activation (Fig. 2I), but RAC1 activation is not sufficient to trigger macropinosome formation in the presence of PTEN (Fig. 2I and K; Supplementary Fig. S3B).
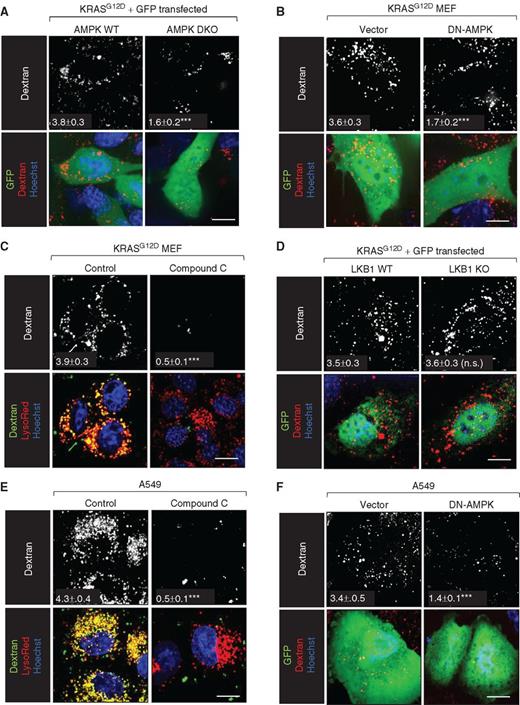
Whether AMPK activation was a general requirement for macropinocytosis or selectively required in PTEN-deficient cells was not clear. The LKB1 tumor suppressor is mutated in up to 30% of non–small cell lung cancer (NSCLC), including tumor cells with activating mutations in KRAS (37). LKB1 is reported to be the major AMPK-activating kinase under metabolic stress (38). It was therefore of interest to determine whether LKB1 and AMPK are required for RAS-driven macropinocytosis. Introduction of KRASG12D drove macropinocytosis in AMPK WT but not AMPK DKO MEFs (Fig. 3A). Consistent with these results, DN-AMPK expression or compound C blocked dextran uptake in KRASG12D MEFs (Fig. 3B and C). Thus, AMPK activity is also necessary for KRAS-driven macropinocytosis. In contrast, KRASG12D expression stimulated equally robust macropinocytosis in LKB1 WT and KO MEFs (Fig. 3D), consistent with studies showing that LKB1 loss reduces but does not eliminate AMPK activity. Moreover, the LKB1-deficient, KRASG12S-expressing NSCLC cell line A549 exhibited a high macropinocytic index that was dramatically reduced by compound C or DN-AMPK expression (Fig. 3E and F). These results suggest that AMPK activation is a general requirement for macropinosome formation.
PTEN-Deficient Prostate Cancer Cells Exhibit Constitutive Macropinocytosis
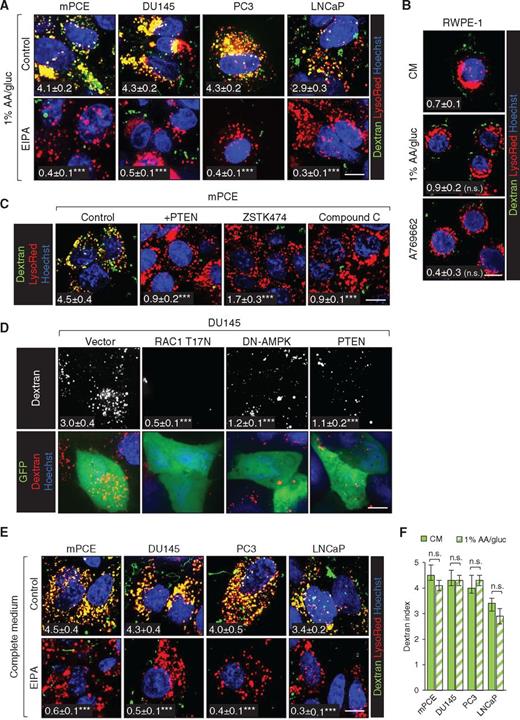
At diagnosis, the majority of prostate tumors exhibit PTEN deficiency or mutation, and complete loss of PTEN is closely linked to the castration resistance and metastasis that render prostate cancer a lethal disease (16). Our results in MEFs suggest that macropinocytosis may supply PTEN-deficient prostate cancers with fuel for biosynthesis and growth. For initial in vitro studies, human prostate cancer cells with PTEN deletion (PC3, LNCaP) or deficiency (DU145) and mouse prostate cancer epithelial (mPCE) cells derived from a tumor in a Ptenflox/flox;Trp53flox/flox;PB-Cre4 mouse, an established model for castration-resistant prostate cancer (CRPC), were utilized (18, 39). All of these PTEN-deficient prostate cancer cell lines exhibited robust, EIPA-sensitive dextran uptake in 1% AA/gluc medium (Fig. 4A). In contrast, the immortalized but nontransformed PTEN-replete prostate epithelial cell line RWPE-1 did not exhibit macropinocytosis under nutrient deprivation or in the presence of A769662 (Fig. 4B). These results suggest that prostatic epithelial cells are not normally macropinocytic and that macropinocytosis is a cancer-associated phenotype that stems from the loss of PTEN function. In keeping with this model, PTEN reconstitution or the pan-PI3K inhibitor ZSTK474 blocked dextran uptake in prostate cancer cells (Fig. 4C and D). As observed in MEFs (Fig. 2C, 3A–C; Supplementary Fig. S3E–S3F), AMPK activation was also necessary for macropinocytosis in prostate cancer cells, as compound C or expression of DN-AMPK blocked macropinocytosis to a similar extent as RAC inhibition (Fig. 4C and D; Supplementary Fig. S6A). Given this requirement for AMPK activation, it was somewhat surprising that prostate cancer cells exhibited equally robust dextran uptake in complete and nutrient-deficient media (Fig. 4E and F). Neither nutrient deprivation nor A769662 increased the amount of dextran taken up by prostate cancer cells over 30 minutes, a time point when steady state was reached (Fig. 4F; Supplementary Fig. S6B and S6C). However, when the dextran pulse was shortened to 5 minutes, A769662 increased uptake in both PC3 and LNCaP prostate cancer cells (Fig. 4G; Supplementary Fig. S6D). These results are consistent with studies demonstrating that AMPK activity is basally elevated in prostate tumors relative to normal tissue (40). AMPK also stimulates autophagy, and blocking macropinocytosis in prostate cancer cells did not further increase autophagic flux (Supplementary Fig. S6E). Together, these results confirm that PTEN loss and AMPK activation promote macropinosome formation in prostate cancer cells even in complete medium.
Albumin Uptake Is Independent of Macropinocytosis in Prostate Cancer Cells
Having established that PTEN-deficient prostate cancer cells exhibit robust macropinocytosis (Fig. 4), whether prostate cancer cells could use macropinocytosis to support growth and survival in low nutrients was evaluated. BSA is routinely used as a fuel in assays designed to measure whether macropinocytosis supports proliferation and survival in low nutrients (6–8). However, BSA is taken up by multiple mechanisms (41). Efficient BSA uptake in MEFs required macropinocytosis (Fig. 1A and C), and thus this cargo could be used to dissect the role of macropinocytosis in resistance to nutrient stress in MEFs (Figs. 1G–I and 2A, D, G, H). In contrast, prostate cancer cells took up similar amounts of BSA in the presence or absence of macropinocytosis (Supplementary Fig. S7A–S7C). As expected, LDL and transferrin, two classic RME cargos, were taken up with equal efficiency in the presence or absence of EIPA, suggesting that BSA entry via RME would also be EIPA resistant (Supplementary Fig. S7D, S7E). Consistent with this model, dominant-negative dynamin1K44A expression inhibited uptake of transferrin and BSA, but not dextran (Supplementary Fig. S7F); RME but not macropinocytosis is dynamin dependent (5). Because these experiments indicated that albumin efficiently enters prostate cancer cells through macropinocytosis-independent pathways, an alternative, physiologically relevant and purely macropinocytic cargo was sought to determine whether macropinocytosis can support prostate cancer anabolism.
Necrotic Cell Debris Is Taken Up by Prostate Cancer Cells Solely by Macropinocytosis
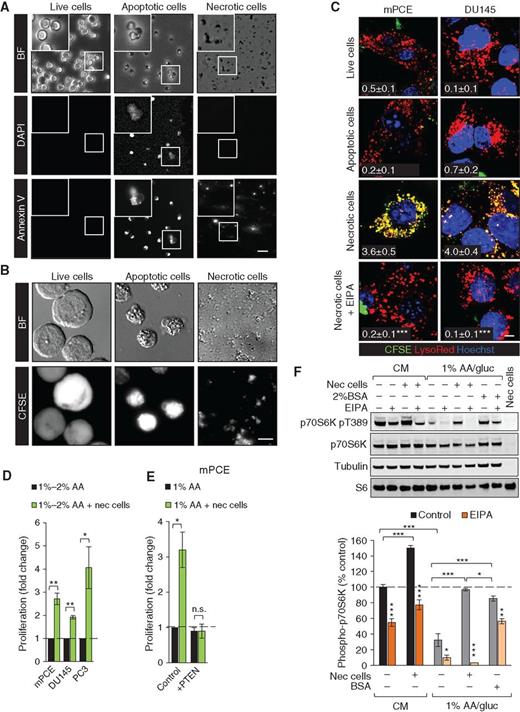
As tumors grow, tortuous and poorly formed vasculature leads to necrosis in regions where oxygen and nutrient delivery are inadequate to meet tumor cell demand (2). Necrosis is present in many aggressive, high-grade tumors, including prostate cancers and RAS-driven tumors, and correlates with negative patient outcomes and resistance to radiation and chemotherapy (42–48). Macropinosomes are large structures, ranging from 0.2 to 5 μm in diameter. Necrotic cell debris could be small enough to be engulfed via macropinocytosis, whereas live or apoptotic cells should be too large to enter via this mechanism. To test this idea, murine hematopoietic FL5.12 cells were fluorescently labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and then killed using different protocols. FL5.12 cells were ideal for these studies because apoptotic death upon withdrawal of the cytokine IL3 avoids the need to remove a cytotoxic drug before supplying the corpses as fuel for prostate cancer cells. A necrotic FL5.12 cell preparation was prepared by allowing sufficient time after IL3 withdrawal for secondary necrosis to occur (48–72 hours). As shown in Fig. 5A and B, freshly killed apoptotic FL5.12 cells are intact, Annexin V– and CFSE-positive, and labeled with the fluorescent vital dye DAPI. In contrast, necrotic FL5.12 cell preparations contain only cell fragments that retain CFSE and Annexin V positivity. As expected, live FL5.12 cells are CFSE-positive but do not stain with DAPI or Annexin V. Consistent with their relative sizes, CFSE-labeled necrotic debris but not apoptotic or live FL5.12 cells were engulfed by macropinocytic mPCE and DU145 cells (Fig. 5C). Importantly, uptake of necrotic debris was fully EIPA-sensitive and observed only in macropinocytic cells, and green necrotic debris fully colocalized with red dextran in macropinosomes (Fig. 5C; Supplementary Fig. S8A and S8B). The selective uptake of necrotic debris but not intact cell corpses or live cells clearly differentiates this process from efferocytosis (phagocytosis of apoptotic cells; ref. 49) or entosis (engulfment of viable cells by cancer cells; ref. 50). To confirm that necrotic debris could be accommodated within macropinosomes, colabeling studies were performed. When prostate cancer cells were fed both red and green dextrans, macropinosomes were uniformly yellow in the merged image as expected (Supplementary Fig. S8C and S8D). In contrast, when red and green necrotic debris were added simultaneously, macropinosomes were either red or green, demonstrating that large cell fragments in the necrotic cell preparation were taken up via macropinocytosis (Supplementary Fig. S8D and S8E). Taken together, these experiments demonstrate that necrotic debris enters cells solely via macropinocytosis.
Whether macropinocytosis could drive prostate cancer cell proliferation in low-nutrient medium was measured using necrotic debris as fuel. Because macropinocytosis in prostate cancer cells did not depend on glucose depletion (Fig. 4E and F), only amino acids were limited to allow maximal macropinocytosis-driven proliferation. The addition of necrotic cell debris significantly stimulated the proliferation of mPCE, PC3, and DU145 cells in amino acid–deficient medium (Fig. 5D). Macropinocytosis was required to derive a benefit from necrotic debris, as PTEN reconstitution eliminated both macropinocytosis (Fig. 4C) and the enhanced prostate cancer cell proliferation (Fig. 5E). Necrotic debris also supported the proliferation of nutrient-deprived macropinocytic KRASG12D MEFs and PANC-1 pancreatic cancer cells but not nonmacropinocytic LSL MEFs or BxPC3 pancreatic cancer cells (Supplementary Fig. S8F and S8G). In fact, adding necrotic debris increased death in nutrient-deprived nonmacropinocytic LSL MEFs or BxPC3 cells, confirming that the necrotic cell preparation did not contain sufficient soluble amino acids to support growth. In summary, macropinocytic cells can use necrotic cell debris to support anabolism under nutrient stress.
To confirm that macropinocytosed material fueled prostate cancer anabolism, we assessed amino acid–sensitive mTORC1 signaling. Amino acids produced by the degradation of macropinosomes can reactivate mTORC1 (7). As expected, shifting prostate cancer cells to amino acid–deficient medium reduced mTORC1-dependent phosphorylation of p70S6 kinase at Thr389 (Fig. 5F). Supplementation with necrotic debris at a concentration of 0.05% protein restored mTORC1 signaling in an EIPA-sensitive manner. BSA supplementation (2%) also restored Thr389 phosphorylation, but this effect was incompletely reversed by EIPA as expected given that BSA uptake is macropinocytosis independent in prostate cancer cells (Fig. 5F; Supplementary Fig. S7A and S7B). Interestingly, necrotic debris increased mTORC1 signaling in an EIPA-sensitive manner even in complete medium, suggesting that macropinocytosis also fuels prostate cancer growth in nutrient-replete conditions (Fig. 5F). Importantly, necrotic cell debris did not itself contribute to the pThr389 p70S6K signal. These results suggest that necrotic debris supplies amino acids to nutrient-deprived macropinocytic prostate cancer cells.
To directly test whether proteins scavenged via macropinocytosis are broken down into amino acids to build biomass, we developed a novel isotopic labeling strategy (Fig. 6A). To label the macropinocytic cargo, FL5.12 cells were grown in stable isotope labeling with amino acids in cell culture (SILAC) medium containing 13C3, 15N1 lysine (K4) and 13C6, 15N4 arginine (R10) for more than 10 generations. Complete labeling of the FL5.12 proteome was confirmed by LC/MS-MS. Necrotic debris containing only fully labeled (K4R10, heavy, 14 Da mass shift) peptides, depicted in green in Fig. 6A–C, was produced from these FL5.12 cells and fed to macropinocytic prostate cancer cells whose proteins contained only unlabeled amino acids (K0R0, light, depicted in blue). Proliferation assays were conducted in medium containing only unlabeled amino acids, and thus the only source of isotopically labeled amino acids was the proteins in the necrotic debris. When prostate cancer cells fed necrotic debris are harvested and their proteins digested with LysC, peptides that contain both lysine and arginine can be present in four different isotopic forms (Fig. 6A–C). Peptides produced independently of macropinocytosis will be unlabeled (K0R0, light, blue). Peptides that contain one unlabeled and one labeled amino acid (K4R0 producing a 4 Da shift or K0R10 producing a 10 Da shift, mixed peaks, red) must have been synthesized in prostate cancer cells using both a labeled amino acid obtained via macropinocytosis and an unlabeled amino acid obtained from the medium. Fully isotopically labeled peptides (K4R10 producing a 14 Da shift, heavy, green) could either come from undigested, heavy-labeled FL5.12 proteins or represent proteins synthesized in prostate cancer cells using both heavy-labeled arginine and lysine obtained via macropinocytosis. An example isotopic profile for the LysC peptide 219FDRGYISPYFINTSK233 from the mitochondrial heat shock protein HSPD1 exhibits these four classes of peptides (Fig. 6C). To summarize, peptides that are K0R0 were generated independent of macropinocytosis, K4R0 and K0R10 peptides can only be generated if macropinocytosis supplies amino acids, and peptides that are K4R10 are from either engulfed but undigested FL5.12 proteins or proteins synthesized in prostate cancer cells using two labeled amino acids derived from macropinocytosis.
To calculate the maximum biomass derived from macropinocytosis, we assumed that all K4R10 peptides (heavy, green) were synthesized in prostate cancer cells using lysine and arginine derived from macropinocytosis (see Supplementary Methods for details). The minimum amount of protein biomass derived from macropinocytosis was calculated by assuming all of these K4R10 peptides were derived from engulfed but undigested FL5.12 proteins. Using this approach, we found that in amino acid–deficient medium, at least 35% to 37% of proteins and as much as 60% to 71% of prostate cancer cell protein biomass was derived from macropinocytosed protein (Fig. 6D and E; Supplementary Fig. S9A). Moreover, even in complete medium where free, unlabeled amino acids were abundant, between 14% and 25% of the prostate cancer cell protein biomass was derived from macropinocytosed protein. To complement the LysC approach, we performed tryptic digests of proteins isolated from human DU145 prostate cancer cells fed heavy-labeled necrotic debris generated from murine FL5.12 cells. In this scenario, uniquely human peptides that contain an isotopically labeled amino acid (K4 or R10) must have been synthesized using amino acids acquired via macropinocytosis. This approach produced similar estimates of the contribution of macropinocytosis to the amino acid pool for protein synthesis (Supplementary Fig. S9B). In summary, isotopic labeling conclusively demonstrates that macropinocytosed protein contributes to prostate cancer biomass even in nutrient-replete conditions.
Cellular corpses are rich sources of other building blocks besides amino acids. Lipids and cholesterol in particular are key drivers of prostate cancer growth (51, 52). Fatty acids and cholesterol are stored in lipid droplets, and depleting these lipid pools suppresses prostate cancer proliferation (51–53). Lysosomal degradation of lipid droplets increases in response to amino acid or glucose depletion as triglycerides in these droplets are utilized to fuel mitochondrial metabolism (53, 54). Using label-free Coherent Anti-stokes Raman Spectroscopy (CARS) to detect C–H stretching vibration signals from lipids (52), lipid droplet content declined significantly in glucose- and amino acid–restricted prostate cancer cells as expected (Fig. 6F; Supplementary Fig. S9C). Supplying necrotic cell debris completely restored lipid droplet content in an EIPA-sensitive manner. As noted earlier (Supplementary Fig. S7D), EIPA did not interfere with LDL uptake, and it did not decrease lipid droplet content when added in complete medium (Supplementary Fig. S9D). Interestingly, supplementation with 2% BSA did not restore lipid droplet content in nutrient-deprived prostate cancer cells (Fig. 6F). This suggests that the membranes and lipids present in necrotic debris rather than proteins are responsible for maintaining lipid stores and that prostate cancer cells can scavenge multiple nutrients from cell corpses.
Prostate Cancer Cells Exhibit Macropinocytosis under Physiologic Conditions
To assess whether normal and transformed prostate epithelial cells perform macropinocytosis under more physiologic conditions, we evaluated dextran uptake in prostate organoids derived from C57BL/6 mice and from Ptenflox/flox;Trp53flox/flox;PB-Cre4 mice that develop autochthonous prostate tumors. PTEN-deficient prostate cancer spheroids but not PTEN-replete normal prostate epithelial organoids exhibited EIPA-sensitive macropinocytosis in nutrient-replete standard 3-D culture medium (Fig. 7A; ref. 55). Macropinocytosis in prostate cancer organoids was sensitive to compound C, suggesting that AMPK activation was also required in this context (Supplementary Fig. S10A). Primary, metastatic tumor cells from patients with CRPC can also be propagated in 3-D culture where they form tumor organoids that exhibit histologic features that mimic the original patient tumor (56). A patient sample deficient in both PTEN and p53, MSK-PCa1, also exhibited constitutive macropinocytosis in 3-D culture (Fig. 7B). Patient-derived xenografts (PDX) were also evaluated in situ. Subcutaneous PTEN- and p53-deficient PDX prostate tumors (Jackson Laboratory PDX model TM00298) also exhibited EIPA-sensitive dextran uptake in vivo following intratumoral injection of dextran (Fig. 7C). Similarly, autochthonous tumors in Ptenflox/flox;Trp53flox/flox;PB-Cre4 mice exhibited robust macropinocytosis of intravenously delivered 70 kD FITC-Ficoll; uptake was again sensitive to systemic EIPA administration (Fig. 7D). Macropinocytosis was not observed in the prostates of normal mice given 70 kD FITC-Ficoll intravenously (Fig. 7E). Importantly, in all cases where macropinocytic uptake was not detected (e.g., in EIPA-treated and wild-type animals), Evans Blue dye that was coinjected with the dextran or Ficoll was present in the tumor or tissue, confirming successful delivery of dextran or Ficoll and EIPA. These results demonstrate that PTEN-deficient prostate cancer cells, but not normal prostate epithelial cells, exhibit robust macropinocytosis in 3-D culture and in vivo.
To evaluate whether prostate tumors rely on macropinocytosis for growth, C57BL/6 mice bearing subcutaneous prostate cancer isografts were treated with EIPA. Consistent with a role for macropinocytosis in driving prostate tumor growth in vivo, alternate day subcutaneous injections of EIPA inhibited both 70 kD FITC-Ficoll uptake and prostate tumor growth, even producing some regressions, without affecting body weight (Fig. 7F–H; Supplementary Fig. S10B–S10E). Again, Evans Blue dye coinjected with Ficoll robustly labeled tumors that failed to take up FITC-Ficoll in the presence of EIPA, confirming successful i.v. injection and that EIPA delivered with this dosing protocol inhibits macropinocytosis (Supplementary Fig. S10E). Although all prostate tumor cells we evaluated exhibited macropinocytosis, xenograft growth of a nonmacropinocytic pancreatic cancer cell line is not affected by EIPA (6). Taken together, these results in cell lines, organoid cultures, and mouse models support the conclusion that prostate cancers use macropinocytosis to acquire extracellular nutrients to fuel growth and proliferation both in vitro and in vivo.
Discussion
This study defines macropinocytosis as a previously unrecognized fuel source in prostate cancer, a tumor class known for its enigmatic nutrient dependencies. This discovery could lead to new therapeutic approaches to this lethal disease. Despite the introduction of second-generation androgen inhibitors such as enzalutamide and abiraterone acetate, patients with CRPC still develop resistance to all available targeted drugs; chemotherapy with docetaxel affords limited clinical benefit and produces significant toxicity (57, 58). Our finding that prostate cancer cells use macropinocytosis to support anabolism suggests that inhibiting this pathway could provide a novel, safe, and effective strategy to target metabolism in late-stage prostate cancer. Macropinocytosis inhibition could synergize with inhibitors of androgen signaling. Androgen deprivation therapy triggers tumor cell death, but castration-resistant disease eventually emerges. The growth of PTEN-deficient tumor cells that survive androgen deprivation therapy may be fueled in part by the macropinocytic catabolism of the corpses of their deceased, androgen-dependent brethren. Similarly, cell death resulting from chemotherapy or radiation therapy may paradoxically increase the nutrient pool available to the surviving PTEN-deficient tumor cells. Inhibitors of lysosomal function such as chloroquine that are often used as autophagy inhibitors may limit prostate cancer cell growth in part by blocking macropinosome degradation. The synthetic sphingolipid SH-BC-893 works upstream from chloroquine, preventing macropinosome–lysosome fusion; SH-BC-893′s antineoplastic activity in prostate cancer models may stem in part from its ability to block macropinocytosis (18). Although EIPA is generally considered to have poor pharmacologic properties, it inhibited both macropinocytosis and tumor growth in mice following systemic administration without obvious toxicity (Fig. 7F–H; Supplementary Fig. S10B–S10E). It will be important to evaluate the sensitivity of autochthonous and metastatic prostate tumors to macropinocytosis inhibitors alone and in combination with standard-of-care therapies in future studies to provide a more complete picture of their potential clinical value.
The discovery that AMPK activation is a general requirement for macropinosome formation in cancer cells dramatically extends our understanding of the regulation of this process. By increasing RAC1-GTP levels, AMPK activation stimulates macropinosome formation (Fig. 2I–K; Supplementary Fig. S5). The relatively slow kinetics of RAC1 activation by A769662 (Fig. 2I), although somewhat unexpected, are consistent with prior work (59). Our finding that A769662 activates RAC1-GTP even more efficiently in PTEN WT MEFs than in PTEN KO cells (Fig. 2I) without stimulating dextran uptake (Supplementary Fig. S3B) seems at first contradictory. However, macropinosome maturation leads to RAC1 inactivation (36), and RAC1 may be trapped in the GTP-bound state in PTEN WT MEFs. Closure of macropinosomes in the presence of elevated PIP3 may prevent the accumulation of RAC1-GTP in PTEN KO MEFs. Intriguingly, the RAC GEFs ARHGEF6 and ARHGEF7 may be substrates for AMPK (60). Additional studies will be required for a full mechanistic understanding of how macropinocytosis is regulated by signal transduction cascades.
Whereas previous studies evaluating the role of macropinocytosis in cancer deprived cells of individual or classes of amino acids (6–8), we utilized media that were deficient either in all amino acids or in both amino acids and glucose. It is difficult to accurately model the nutrient stress that tumors will experience in their normal microenvironment, but inadequate perfusion is likely to restrict access to multiple nutrients simultaneously. The 1% nutrient condition was selected because nutrient titration experiments indicated that this was the least severe reduction in nutrients that killed the majority of wild-type MEFs within 48 hours. The specific nutrient conditions used can influence conclusions regarding the importance of macropinocytosis. When PTEN KO MEFs are deprived of only amino acids, they do not exhibit macropinocytosis (Fig. 2B). Amino acid limitation would be relieved only by autophagy, a catabolic process that cannot by itself drive cell-autonomous growth (Fig. 2F). In contrast, combined glucose and amino acid stress (1% AA/gluc) activates AMPK and stimulates macropinocytosis (Fig. 2B), thereby providing amino acids from the degradation of albumin in the medium. That AMPK-driven macropinocytosis is responsible for the growth-stimulatory effects of glucose deprivation is supported by the observations that: (i) A769662 enhances proliferation in macropinocytosis-competent amino acid–deprived PTEN KO MEFs but not nonmacropinocytic PTEN WT MEFs (Fig. 2A), (ii) PAK1 is necessary for A769662 to drive macropinocytosis and proliferation in PTEN-deficient MEFs in low nutrients (Fig. 2F), and (iii) A769662 can drive proliferation in PTEN-deficient MEFs in low nutrients even in the absence of autophagy (Fig. 2H). Thus, it is not glucose limitation per se but rather AMPK-induced macropinocytosis that stimulates proliferation in Fig. 2A. Indeed, direct AMPK activation in full glucose (1% AA medium + A769662) permits almost twice as much proliferation as AMPK activation by glucose limitation (1% AA/gluc). It is worth noting that, although autophagy is not sufficient to fuel proliferation in low-nutrient medium, autophagy is necessary for maximal macropinocytosis-driven proliferation (Fig. 2H), most likely because it spares amino acids derived from macropinocytosis for anabolism. In conclusion, the seemingly paradoxical effect of glucose deprivation on the proliferation of amino acid–deprived MEFs is readily explained by the stimulation of macropinocytosis.
An intriguing implication of this study is that AMPK inhibitors could be deployed against cancer cells as macropinocytosis inhibitors. AMPK is a negative regulator of macromolecular synthesis and often classified as a tumor suppressor based on its ability to limit anabolism and stimulate catabolic processes (25). On the other hand, multiple studies suggest that AMPK plays a supportive role in established tumors similar to the reciprocal role of autophagy as a suppressor of tumor initiation and a driver of tumor progression (38, 61–64). Indeed, many studies indicate that AMPK promotes prostate cancer progression (40). AMPK stimulates autophagy, and autophagy inhibitors have been incorporated into combination therapies based on the premise that blocking the stress response will sensitize tumor cells to other drugs. Like autophagy, macropinocytosis may provide a bypass system that could rescue tumor cells from metabolic stress induced by other agents. Our finding that AMPK promotes not just autophagy but also macropinocytosis suggests that AMPK inhibitors could be effective against macropinocytic tumors alone or as a part of drug combinations.
Histologic evidence of tumor necrosis is a negative prognostic indicator that is correlated with recurrence and aggressive, metastatic disease in multiple solid tumors, including prostate cancer and RAS-driven malignancies (42–46). Our observations that necrotic cell corpses are a rich source of proteins and lipids capable of driving both PTEN-deficient prostate cancer cell and KRAS-driven pancreatic cancer cell growth (Fig. 5D and E; Supplementary Figs. S8F and S8G) may partially explain this correlation. Necrosis may enhance tumor growth by triggering inflammation (65) while at the same time providing a fuel source for the cytokine-driven growth of macropinocytic tumor cells. The correlation between inflammatory markers and necrosis is imperfect (47), and in some tumors necrosis may promote cancer cell anabolism primarily by providing metabolic substrates rather than by stimulating a protumorigenic immune response. It is important to recognize that any extracellular material small enough to fit in a macropinosome could be consumed and catabolized through this pathway; cargoes that are taken up by selective pathways, for example, albumin, would still be taken up nonselectively by macropinocytosis. It is also significant that necrotic cell debris was supplied here at only 0.05% to 0.1% protein, a 20- to 40-fold lower concentration than BSA, suggesting that relatively small amounts of necrosis could have a significant impact on tumor growth. Rather than attempting to define the relative anabolic value of different macropinocytic cargos in vivo, the most critical next step is to uncover more specific ways to disrupt macropinocytosis than the chemical tools currently available in order to better define the importance of this nutrient-scavenging pathway for tumor initiation and progression. Therapeutically, any inhibitor of macropinocytosis would simultaneously block the uptake of BSA, necrotic debris, and any other extracellular cargos. Although the contribution that dead cell catabolism makes to tumor cell growth is difficult to quantify, the uptake of necrotic debris through macropinocytosis (necrocytosis) defines a new way the microenvironment likely supports tumor cell anabolism.
These studies also highlight that caution is required when using BSA as a tool to evaluate the contribution of macropinocytosis to anabolism and growth, as there are significant, cell line–specific differences in the relative amount of BSA that is taken up through macropinocytosis and dynamin-dependent processes like RME (Fig. 1A and B; Supplementary Fig. S7A, S7B, and S7F). In cell lines that take up BSA efficiently in the absence of macropinocytosis (Supplementary Fig. S7A, S7B), necrotic cell debris provides an alternative, physiologically relevant substrate that is taken up solely by macropinocytosis (Fig. 5C). Moreover, necrotic cells are likely a more complete diet than extracellular protein. The observation that necrotic debris, but not extracellular protein, can maintain lipid droplets in nutrient-restricted cells (Fig. 6F) is strong evidence that the membrane component of dead cells can be efficiently recycled via macropinocytosis. From a technical perspective, using necrotic cell debris as macropinocytic cargo has the advantage of facile fluorescent (Fig. 5A–C; Supplementary Fig. S8A-S8E) or isotopic (Fig. 6A–E) labeling, permitting both tracing of the engulfed material and analysis of its ultimate fate. Importantly, the novel SILAC labeling approach we developed to show that macropinocytosed proteins contribute amino acids to protein synthesis in nutrient-replete as well as nutrient-limited media is readily adoptable by other research groups. We anticipate that the use of necrotic cells as cargo will facilitate future studies defining the role of macropinocytosis in tumor cell growth.
In conclusion, although the anabolic value of macropinocytosis in tumor cells is now well accepted, there remains an unmet need for specific agents that block only macropinosome formation or degradation to better define the importance of this process in tumor growth and progression, particularly in vivo. Although their pleiotropic actions somewhat complicate the dissection of the role of macropinocytosis in cancer, NHE inhibitors, AMPK inhibitors, and SH-BC-893 may actually prove to be superior therapeutic agents for the very reason that they possess multifaceted and complementary antineoplastic effects (18).
Methods
Cell Culture
DMEM or RPMI containing 1% of the normal amount of amino acids and/or glucose was generated by preparing DMEM or RPMI lacking amino acids and/or glucose from chemical components and mixing it 99:1 with complete medium. In all experiments, the medium was supplemented with 10% standard fetal calf serum, which supplies amino acids, glucose, and albumin. LNCaP (2016), PC3 (2011), DU145 (2011), A549 (2013), PANC-1 (2013), BxPC3 (2017), and RWPE-1 (2012) were obtained from the ATCC in indicated years. mPCE cells and PTEN WT/KO MEFs were generated in the Edinger lab (2015); other MEF lines were obtained from collaborating labs in 2003 (LSL/KRASG12D), 2007 (AMPK WT/DKO and ATG5 WT/KO), or 2017 (PAK1 WT/KO). Cell lines were cultured for ≤3 weeks, at which point a frozen low-passage (≤4 weeks after receipt) stock was thawed. Cell lines were authenticated by evaluating the expression patterns of androgen receptor (PC3, DU145, LNCaP, and mPCE) and gene deletions (MEFs, LNCaP, PC3, and mPCE) at least every 6 months. Cells were tested for Mycoplasma at least every 4 months (VENOR GeM PCR Kit, Sigma). Before they were used in experiments, PAK1 WT and KO MEFs were cured of Mycoplasma by culture in ciprofloxacin for 2 months (PCR confirmed); all other cell lines tested negative. Additional details in Supplementary Methods.
Light Microscopy
Unless otherwise indicated, fluorescent dextran (1 mg/mL), Alexa 488 BSA (0.5 mg/mL), or Alexa 488 transferrin (0.5 mg/mL) were added in combination with Lysotracker Red (1:10,000 dilution) and Hoechst 33342 for 30 minutes, cells were washed three times with PBS, and live cells evaluated on a spinning disk confocal microscope. Dextran index was calculated using ImageJ software as detailed in ref. 20 and Supplementary Fig. S1A. When nutrient-deficient medium was used, 70 kD dextran or BSA uptake was measured after a 16-hour incubation; cells were preincubated with 50 μmol/L A769662 for 2 hours. EIPA was used at 50 to 75 μmol/L and added 1 hour prior to dextran addition. In Dil-LDL uptake assays, cells were incubated in media with 10% charcoal-stripped serum for 24 hours, then 20 μg/mL Dil-LDL was added ± EIPA for 2 hours.
SILAC Labeling
FL5.12 cells were incubated in SILAC media containing “heavy” 13C- or 15N-labeled arginine (R10) and lysine (K4) for >10 divisions. FL5.12 cells were then killed by IL3 withdrawal; cells were maintained at high density and 72 hours allowed for secondary necrosis to produce necrotic debris. Supernatant was fully aspirated from necrotic cell pellets, which were used directly or stored at −80°C. In isotopic labeling experiments, prostate cancer cells were washed with PBS after 72 hours, lysed, and proteins digested with trypsin or Lys-C and analyzed by mass spectrometry as described in the Supplementary Methods.
RAC1 Activity Assays
MEFs were transfected with biosensors generously provided by the Hahn Laboratory (University of North Carolina): (i) RAC1 FLARE dual-chain biosensor (CyPet-RAC1 and YPet-PBD); (ii) RAC1 constitutively active dual-chain biosensor (CyPet-RAC1-Q61L and YPet-PBD); or (iii) CyPet-RAC1 donor alone. Cells were imaged 24 hours after transfection and stimulated with 50 μmol/L A769662 where indicated. FLIM-FRET measurements of the RAC1 FLARE biosensors were acquired and processed using the SimFCS software package developed at the Laboratory for Fluorescence Dynamics (www.lfd.uci.edu) as described in the Supplementary Methods.
Lipid Droplet Content Measurement by CARS
The CARS imaging system is described in detail in ref. 66. Cells were fixed with 4% formaldehyde and imaged with a 60× water objective. The laser power on the sample was at 10 mW with 10-ms pixel dwell time. The lipid droplet area was estimated from CARS images using a customized Matlab program. Four components Otsu thresholding method was used to separate the lipid droplets, cell cytoplasm, cell nucleus, and the background. The lipid droplet area was defined as the number of pixels covered by lipid droplets over the number of pixels covered by cytoplasm.
In Vivo Experiments
Experiments conducted in mice were performed in accordance with the Institutional Animal Care and Use Committee of the University of California, Irvine. Prostate isografts were produced by injecting 5 million mPCE cells subcutaneously in the flank of 5-week-old C57BL/6 mice. Once tumors reached 100 mm3, animals were randomly assigned to either the vehicle (1% DMSO in PBS) or EIPA (7.5 mg/kg subcutaneously every other day) group (n = 10–11). Tumor volume was calculated using the formula volume (mm3) = length [mm] × (width [mm])2 × 0.52. For in vivo dextran uptake analysis, JAX PDX TM00298 tumors were intratumorally injected with 2 mg Oregon Green dextran dissolved in 1% Evans Blue dye 1 hour after intraperitoneal (i.p.) injection with vehicle or 10 mg/kg EIPA. Ten-week-old male C57BL/6, Ptenflox/flox;Trp53flox/flox;PB-Cre4, or C57BL/6 mice with or without mPCE isografts were intravenously injected with 250 mg/kg FITC-Ficoll dissolved in 1% Evans Blue dye 1 hour after i.p. injection of vehicle or 10 mg/kg EIPA or 1.5 hours after subcutaneous injection of 7.5 mg/kg EIPA as indicated.
Statistical Methods and Data Analysis
Significance was determined using two-tailed t tests. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant (P > 0.05). The Tukey method was used to correct for multiple comparisons.
Disclosure of Potential Conflicts of Interest
A.L. Edinger has ownership interest in a patent filed covering SH-BC-893. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions
Conception and design: S.M. Kim, T.T. Nguyen, A. Ravi, P. Kubiniok, V. Jayashankar, L. Malacrida, B.J. Tromberg, A.L. Edinger
Development of methodology: S.M. Kim, T.T. Nguyen, A. Ravi, P. Kubiniok, B.T. Finicle, M.A. Digman, B.J. Tromberg, A.L. Edinger
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S.M. Kim, T.T. Nguyen, A. Ravi, P. Kubiniok, B.T. Finicle, V. Jayashankar, L. Malacrida, J. Hou, J. Robertson, J. Chernoff, M.A. Digman
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S.M. Kim, T.T. Nguyen, A. Ravi, P. Kubiniok, B.T. Finicle, V. Jayashankar, L. Malacrida, J. Hou, M.A. Digman, P. Thibault, A.L. Edinger
Writing, review, and/or revision of the manuscript: S.M. Kim, T.T. Nguyen, A. Ravi, P. Kubiniok, B.T. Finicle, V. Jayashankar, L. Malacrida, J. Chernoff, M.A. Digman, E.O. Potma, B.J. Tromberg, P. Thibault, A.L. Edinger
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): T.T. Nguyen, A. Ravi, P. Kubiniok, V. Jayashankar, D. Gao, A.L. Edinger
Study supervision: B.J. Tromberg, A.L. Edinger
Acknowledgments
The authors thank Yu Chen (Memorial Sloan Kettering Cancer Center) for generously providing MSK-PCa1 and David Fruman (University of California, Irvine) for comments on the manuscript. This work was supported by grants to A.L. Edinger from the NIH (R01 GM089919), CDMRP (W81XWH-11-1-0535), the American Cancer Society (RSG-11-111-01-CDD), the University of California Cancer Research Coordinating Committee (CRR-17-426826), and UCI Applied Innovation. B.J. Tromberg, E.O. Potma, and J. Hou were supported by NIH/NIBIB Biomedical Technology Research Center Laser Microbeam and Medical Program (LAMMP): P41EB015890. S.M. Kim was supported by GAANNP200A120207. L. Malacrida and M.A. Digman were supported in part by grants NIH-P41-RR03155 and NIH-P50-GM076516. Core facilities at UCI were supported by Cancer Center Support grant P30 CA62203. Proteomics work was supported by Canada Research Chairs in Proteomics and Bioanalytical Mass Spectrometry (P. Thibault), the Genome Canada Genomics Innovation Network (P. Thibault), and the National Science and Engineering Research Council (311598, P. Thibault). P. Kubiniok is supported by a Vanier scholarship.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
Supplementary data
intensity video
GP FRET video



![Figure 6. Necrotic debris consumed by macropinocytosis is used to build biomass. A, Proteomics workflow. Unlabeled mouse mPCE or human DU145 prostate cancer cells were fed K4R10-labeled necrotic FL5.12 cells for 72 hours in 1% AA or complete medium (CM), washed with PBS 3 times, then digested with LysC for LC/MS peptide analysis. B, Hypothetical predicted mass spectra of one peptide from the experiment in A. Light peaks (blue) do not contain lysine or arginine derived from macropinocytosed proteins. Medium peaks (red) correspond to newly synthesized protein containing both unlabeled [macropinocytosis (MP)–independent] and labeled (MP-dependent) lysine and arginine. Heavy peaks (green) correspond to newly synthesized peptides containing both heavy labeled lysine and arginine (MP-dependent) or peptides from engulfed undigested necrotic debris. C, Actual peptide spectra of a triply charged peptide (amino acid sequence FDRGYISPYFNTSK from the HSPD1 protein) from LysC digested mPCE cells fed necrotic debris. D and E, Maximum and minimum protein biomass derived from macropinocytosed debris calculated as depicted graphically using violin plots (D) or described in the text and tabulated in E. F, DU145 cells were cultured for 24 hours in 1% AA/gluc ± necrotic debris, 2% BSA, ± EIPA (50 μmol/L), then lipid droplets were imaged by CARS. Scale bar, 20 μm. See also Supplementary Fig. S9.](https://aacr.silverchair-cdn.com/aacr/content_public/journal/cancerdiscovery/8/7/10.1158_2159-8290.cd-17-1215/6/m_866fig6.jpeg?Expires=1716882334&Signature=3i9Rrur5D8oNW4oxVzTupXLOqU2W3~u8sbsJAPsb5i0zG-F4DPrYcanvep7dZ56XRdrnqvj6WdDqlLb79oTsLDKGU~8YSILfy4ugaV5KnENFd4YYMb80ySTplLvoUDsfOgbVmhaglBEY4GYOWZsqKsO4wj-XuUdoyJmJpxQ5BIwFIFMt9ZkfIvaeOaNfpoWTfBVo57gAacToraMZ63HCFJpq9brtyW5Xfz9abbKEMKsyV5xyaFaPBsnr~F08DKHTMEEBCY7oj8wbM52jRIK3v2fzrxdZ3vLhJcIMAQ4dOnHMBaNn4POrzX4HrDo5NsGJQnygqQvB9hIF0sfc2nxSrg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
