Abstract
Adaptive resistance to MEK inhibitors (MEKi) typically occurs via induction of genes for different receptor tyrosine kinases (RTK) and/or their ligands, even in tumors of the same histotype, making combination strategies challenging. SHP2 (PTPN11) is required for RAS/ERK pathway activation by most RTKs and might provide a common resistance node. We found that combining the SHP2 inhibitor SHP099 with a MEKi inhibited the proliferation of multiple cancer cell lines in vitro. PTPN11 knockdown/MEKi treatment had similar effects, whereas expressing SHP099 binding–defective PTPN11 mutants conferred resistance, demonstrating that SHP099 is on-target. SHP099/trametinib was highly efficacious in xenograft and/or genetically engineered models of KRAS-mutant pancreas, lung, and ovarian cancers and in wild-type RAS-expressing triple-negative breast cancer. SHP099 inhibited activation of KRAS mutants with residual GTPase activity, impeded SOS/RAS/MEK/ERK1/2 reactivation in response to MEKi, and blocked ERK1/2-dependent transcriptional programs. We conclude that SHP099/MEKi combinations could have therapeutic utility in multiple malignancies.
Significance: MEK inhibitors show limited efficacy as single agents, in part because of the rapid development of adaptive resistance. We find that SHP2/MEK inhibitor combinations prevent adaptive resistance in multiple cancer models expressing mutant and wild-type KRAS. Cancer Discov; 8(10); 1237–49. ©2018 AACR.
See related commentary by Torres-Ayuso and Brognard, p. 1210.
This article is highlighted in the In This Issue feature, p. 1195
Introduction
The RAS/ERK pathway is one of the most commonly affected signaling pathways in human cancer (1–3). Mutations in genes encoding pathway components, including those for receptor tyrosine kinases (RTK), SHP2, NF1, RAS, or RAF, cause inappropriate pathway activation and promote oncogenesis. Attempts have been made to target the ERK pathway in different cancer types and can lead to initial responses. Unfortunately, a form of intrinsic resistance termed “adaptive resistance” occurs frequently, resulting in lack of efficacy, recurrence, or progression (4).
KRAS is the most frequently mutated RAS/ERK pathway gene (1–3). Approaches to target KRAS-mutant cancers with MEK inhibitors (MEKi) have failed, often due to the induction of RTK genes and/or their ligands. For example, FGFR1 is activated in MEKi-treated KRAS-mutant lung cancers, leading to increased upstream signaling and ERK reactivation (5). Another group found that MEKi resistance can be mediated through ERBB3 in KRAS-mutant lung and colon cancers (6), whereas a third reported that MEKi treatment leads to EGFR activation in KRAS-mutant pancreatic cancer lines (7). Malignancies that lack mutations in pathway genes but nonetheless hyperactivate ERK also show adaptive resistance in response to MEKi. For example, MEKi-treated triple-negative breast cancer (TNBC) cells induce the expression of genes encoding AXL, DDR1, FGFR2, IGF1R, KIT, PDGFRB, and VEGFRB (8, 9).
Because resistance to MEKi can be mediated by multiple RTKs, combining MEK and RTK inhibition is probably not a viable therapeutic approach. However, a strategy that efficiently blocks signals from multiple activated RTKs might prevent adaptive resistance. The protein-tyrosine phosphatase SHP2 is a positive (i.e., signal-enhancing) signal transducer, acting between RTKs and RAS (10, 11). A potent, highly specific inhibitor targeting SHP2, SHP099, has been developed and blocks ERK activation and proliferation of cancer cells driven by overexpressed, hyperactivated RTKs (12, 13). We hypothesized that SHP099 would inhibit signals from RTKs activated following MEK inhibition and thereby block adaptive resistance. This idea comports with the previous finding that PTPN11 shRNA or CRISPR/Cas9-mediated deletion prevents adaptive resistance to vemurafenib in BRAF-mutant colon cancer (14).
Here, we test this hypothesis in multiple KRAS-mutant and wild-type (WT) cancer cells from different histotypes. Our results suggest that SHP2 inhibition could provide a general strategy for preventing MEKi resistance in a wide range of malignancies and might also have single-agent efficacy against KRAS mutants that retain significant GTPase activity.
Results
SHP099 Abrogates Adaptive Resistance to MEKi In Vitro
Previous work showed that several cancer models develop adaptive resistance to MEKi through RTK upregulation. We analyzed RTK/RTK ligand gene expression by qRT-PCR in pancreatic ductal adenocarcinoma (PDAC) cell lines treated with AZD6244, a well-established MEKi (Fig. 1A). Consistent with earlier findings, several—but different—RTKs were induced by MEKi treatment, including EGFR, FGFR3, IGFR1, MET, and PDGFRB in MIAPaCa-2 cells, ERBB2/3, FGFR2/3, and IGFR1 in Capan-2 cells, and ERBB2/3 and FGFR3 in CFPAC-1 cells. The same lines variably induced EGF, FGF2, PDGFB, PDGFC, PDGFD, and/or VEGFA/B. These observations make it difficult, if not impossible, to design an efficient combination therapy with MEKi by targeting RTKs directly.
To explore whether SHP2 inhibition could suppress MEKi adaptive resistance, we performed in vitro viability (PrestoBlue) and colony formation assays on a panel of KRAS-mutant PDAC lines (Fig. 1B and C). Resistant cell populations and drug-resistant colonies were observed after 7 or 10 days, respectively, of AZD6244 treatment. AZD6244 itself had variable effects, leading to 30% to 90% reduction in proliferation/colony formation compared with control DMSO treatment; nevertheless, nearly all lines showed significant resistance. Consistent with a previous report (12), KRAS-mutant cell lines exhibited low sensitivity to SHP099 alone. By contrast, all but two of the lines had markedly reduced cell numbers and few or no detectable colonies after SHP099/MEKi combination treatment; in most cases, the combination was synergistic (Fig. 1B, red asterisks; Supplementary Table S1). Similar effects were seen in growth curve assays (Supplementary Fig. S1A), with the more potent MEKi trametinib (Supplementary Fig. S1B), and on short-term cultures of cells from patient-derived xenografts (PDX) and in KRAS-mutant non–small cell lung cancer (NSCLC) lines (Supplementary Fig. S1C–S1E). The drug combination decreased cell-cycle progression and, in some lines, enhanced cell death (measured at 48 hours and 6 days of treatment, respectively), compared with either single agent alone (Supplementary Fig. S1F).
Expression of a mutant (PTPN11P491Q) predicted to lack SHP099 binding in MIAPaCa-2 cells (which are quite sensitive to SHP099/MEKi) and in KPC 1203, a cell line derived from induced LSL-KRASG12DTrp53R172H (KPC) mice (15), eliminated the effects of SHP099 in combination-treated cells (Fig. 1D and E). Another drug-resistant mutant, PTPN11T253M/Q257L (12), rescued the effects of the combination on H358 NSCLC cells (Fig. 1D). Moreover, combining MEK inhibition and PTPN11 shRNA expression had similar effects to SHP099/MEKi treatment (Fig. 1F). These data indicate that SHP099 is “on-target” and that SHP2 inhibition diminishes adaptive resistance to MEKi in multiple KRAS-mutant cancer cell lines, arising from two distinct tissues.
SHP099 Impedes MEKi–Induced Reactivation of the MAPK Pathway
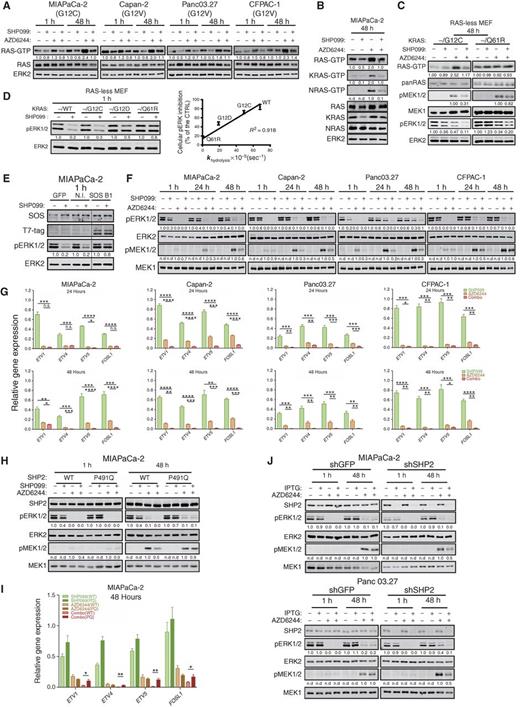
We assessed the biochemical effects of each single agent and the drug combination on RAS/ERK pathway activity after short-term (1 hour) and longer-term (48 hours) treatments. Short-term AZD6244 exposure had no detectable effect on RAS. At 48 hours, however, RAS activation (monitored by RAF-RBD assay) was enhanced, consistent with signaling from the induced RTKs/RTK ligands (Fig. 2A). Isoform-specific antibodies revealed increased activation of KRAS and NRAS in response to 48 hours MEKi treatment of MIAPaCa-2 cells (Fig. 2B). SHP2 acts upstream of RAS, but whether it promotes RAS exchange (e.g., via SOS), inhibits RAS-GAP, or both, has been less clear (10, 11). MIAPaCa-2 cells have no WT KRAS (mutant allele frequency = 0.99; refs. 16, 17), so the increase in KRAS activation following MEKi treatment must reflect enhanced cycling of KRASG12C. As KRASG12C is highly resistant to RAS-GAP (18), the decreased KRAS-GTP in SHP099/AZD6244-treated MIAPaCa-2 cells indicates that residual KRASG12C GTPase activity contributes significantly to the steady-state level of KRAS-GTP in these cells and that SHP2 must promote RAS exchange. Increased NRAS-GTP in response to SHP099 reflects activation of normal, endogenous NRAS.
The other PDAC lines tested express KRAS mutants with less intrinsic GTPase activity than KRASG12C (18) and retain WT KRAS. Hence, it was not clear whether SHP099 can also block activation of these RAS mutants in response to MEKi treatment or affects WT KRAS or the other RAS isoforms (Fig. 2A). To more directly interrogate the effects of SHP2 inhibition on other KRAS mutants, we used RAS-less mouse embryonic fibroblasts (MEF; ref. 19). As in MIAPaCa-2 cells, KRASG12C-reconstituted RAS-less cells showed increased KRAS-GTP after 48 hours of MEKi treatment, and this increase was prevented by SHP099. By contrast, SHP099 had no effect on KRASQ61R-GTP levels (Fig. 2C). The ability of single-agent SHP099 to inhibit ERK activation in RAS-less MEFs reconstituted with different KRAS mutants was linearly related to their reported GTPase activities (ref. 17; Fig. 2D). These results confirm that SHP2 is required for RAS exchange, most likely acting upstream of SOS1/2. Indeed, expressing the SOS1 catalytic domain tagged with a C-terminal CAAX BOX of RAS (20) rescued the effects of SHP099 on ERK activation in MIAPaCa-2 cells (Fig. 2E).
Single-agent AZD6244 blocked MEK activity and ERK1/2 phosphorylation after 1 hour, but these effects were successively abolished after 24 and 48 hours of treatment, respectively, and MEK and ERK activity rebounded (Fig. 2F; Supplementary Fig. S2A). Trametinib also caused MEK/ERK rebound, although to a lesser extent (Supplementary Fig. S2B). Consistent with its effects on RAS, SHP099 coadministration blocked the adaptive increase in MEK and ERK phosphorylation in response to either MEKi (Fig. 2F; Supplementary Fig. S2A and S2B). ERK-dependent gene expression can provide a more sensitive assessment of pathway output than phospho-ERK (pERK) levels (21), so we measured FOS-like 1 (FOSL1) and ETS variants 1, 4, 5 (ETV1, 4, and 5) RNA by qRT-PCR. Compared with the effects of either single agent, the SHP099/AZD6244 or SHP099/trametinib combination caused greater suppression of ERK-dependent transcription (Fig. 2G; Supplementary Fig. S2C). Other RTK-evoked pathways (e.g., PI3K/AKT, STAT, and JNK/p38) showed no consistent effects of either single agent or the drug combination (Supplementary Fig. S2D and data not shown). These findings confirm that ERK reactivation is a key component of the adaptive program activated in KRAS-mutant cancer cells treated with MEKi and show that SHP099 blocks this adaptive response. Importantly, the biochemical effects of SHP099 (like its effects on colony formation; Fig. 1D) were reversed in MIAPaCa-2 (Fig. 2H), H358 (Supplementary Fig. S2E), and KPC 1203 cells expressing SHP099-resistant SHP2. PTPN11 depletion had similar biochemical effects as SHP2 inhibition (Fig. 2J), confirming on-target effects of SHP099.
We also explored the mechanism of resistance of two KRAS-mutant PDAC lines to SHP099/MEKi. PSN1 cells failed to suppress MEK–ERK reactivation or ERK-dependent gene expression (Supplementary Fig. S3A and S3B). In SU.86.86 cells, MEK/ERK and ERK-dependent genes were inhibited to an extent similar to sensitive cells, consistent with a downstream escape mechanism (Supplementary Fig. S3C and S3D). Further investigation will be required to uncover the precise molecular explanation for resistance in these cell lines.
Combined SHP2/MEK Inhibition Suppresses KRAS-Mutant Tumor Growth In Vivo
We next established Capan-2, MIAPaCa-2, and H358 xenografts and treated them with vehicle control (methyl-cellulose+Tween80), trametinib alone, SHP099 alone, or SHP099/trametinib. We used trametinib because of its favorable mouse pharmacokinetic properties (t½ = 33 hours; ref. 22), which enable single daily dosing, as does SHP099 (13). In initial experiments, mice were treated daily with trametinib (1 mg/kg), a dose used commonly in mouse tumor studies (6, 23, 24), SHP099 (75 mg/kg), or both. Each single agent was well tolerated, but mice receiving the combination lost weight (>10%), exhibited lassitude, and began dying at day 7 of treatment. Some showed gross gastrointestinal (GI) bleeding (Supplementary Fig. S4A), and histology revealed multifocal GI tract ulceration, acute esophagitis and gastritis, and villus blunting, which could explain malabsorption, diarrhea, and weight loss (Supplementary Fig. S4B).
Although trametinib is usually administered to mice at this dose or even at doses as high as 3 mg/kg (5, 25), the mouse allometric equivalent of the maximum tolerated dose (MTD) in humans is ∼0.25 mg/kg (26). We treated a small group of mice with this dose of trametinib and SHP099 (75 mg/kg) daily (q.d.). Although treated mice lived longer than with the higher trametinib dose, this combination also led to weight loss and death (data not shown). Exploratory dose finding resulted in a tolerable schedule, in which trametinib is delivered at 0.25 mg/kg, with SHP099 (75 mg/kg) every other day (q.o.d.). These mice showed no observable histopathology (Supplementary Fig. S4C). A few developed mild, self-limited, non-bloody diarrhea, but all showed stable weight and normal behavior and appeared healthy for up to 37 days of continuous treatment (Supplementary Fig. S4D and S4E and data not shown).
Capan-2, MIAPaCa-2, or H358 xenografts were allowed to grow to 500 mm3 (Fig. 3A), and then mice were treated with vehicle, trametinib (0.25 mg/kg q.d.), SHP099 (75 mg/kg q.d.), or trametinib (0.25 mg/kg q.d.)/SHP099 (75 mg/kg q.o.d.). Remarkably, the combination caused substantial regressions in all mice (Fig. 3A; Supplementary Fig. S4F). Tumor shrinkage averaged >65% in the three models, well above RECIST criteria (27). The decrease in tumor size probably underestimates the antineoplastic effect, as the ratio of tumor cells/area also decreased, with the residual area occupied by fibroblasts (Fig. 3B and C). Strikingly, Capan-2 xenografts treated for 37 days with SHP099/trametinib failed to regrow after 40 days of drug withdrawal (Fig. 3D). The residual tumor appeared to have undergone cellular senescence, as shown by βgal staining (Fig. 3E) and elevated expression of the senescence-associated cytokine IL6 (Fig. 3F).
Single-agent effects were more variable both within each treatment group and in mice bearing MIAPaCa-2 versus Capan-2/H358–derived tumors. Trametinib had minimal effects on MIAPaCa-2 tumors, although it caused significant shrinkage of about half of the Capan-2 and H358 xenografts (Fig. 3A; Supplementary Fig. S4F). Only a few trametinib-treated mice met RECIST criteria (>30%), however, and the SHP099/MEKi combination was more effective (Fig. 3A; Supplementary Fig. S4F). Surprisingly, in contrast to its lack of effect on proliferation in cell culture or on colony formation, SHP099 alone caused tumor shrinkage in ∼80% of Capan-2, ∼60% of MIAPaCa-2, and ∼70% of H358 xenografts. It is not clear whether this discrepancy reflects effects of SHP099 on normal RAS/ERK signaling in cells within the tumor microenvironment (e.g., fibroblasts and blood vessels), effects on the malignant cells themselves with secondary consequences for stroma, or both. Consistent with at least some nonautonomous effects, SHP099 decreased tumor vascularity as monitored by CD31 immunostaining (Fig. 3B and C), without major effects on VEGF or FGF mRNA levels (see Fig. 3J). Nevertheless, single-agent SHP099 was, like trametinib alone, inferior to the drug combination (Fig. 3A; Supplementary Fig. S4F). Comporting with these biological effects, immunoblotting (Fig. 3G; Supplementary Fig. S4G), qRT-PCR (Fig. 3H), and IHC analysis (Fig. 3I) revealed greater pERK inhibition in combination- than in single agent–treated tumors. Notably, tumors induced RTK and RTK ligand expression following MEKi treatment, confirming that adaptive resistance via RTK overactivation occurs in vivo (Fig. 3J).
We also tested syngeneic mice injected orthotopically with KPC 1203 cells. Tumors were allowed to grow for 10 days, 5 mice were sacrificed to obtain baseline tumor sizes, and the rest were treated with single agent or SHP099/trametinib for 5 or 15 days, respectively. Again, mice in the combination arms showed markedly inhibited tumor growth, compared with trametinib-treated mice (Fig. 3K and L). Although the combination was superior (day 15; Fig. 3L), SHP099 also had significant effects, even though KRASG12D has significantly less residual GTPase activity than KRASG12C (18). Immunoblot analysis revealed greater inhibition of pERK and DUSP6 (an ERK target gene product) in combination-treated tumors than in those treated with trametinib or SHP099 alone (Fig. 3M).
As in the xenografts, tumor vascularity and overall tumor cellularity was reduced in combination-treated genetically engineered mouse models (GEMM; Fig. 3N and O). Furthermore, residual tumor cells in combination-treated mice showed ductal differentiation, compared with vehicle- or single agent–treated mice (Fig. 3P). PAS/Alcian Blue staining revealed secretory activity (Fig. 3P), whereas qRT-PCR analysis showed induction of ductal and, more prominently, endocrine markers (Supplementary Fig. S4H). All SHP099/trametinib-treated xenografts and syngeneic tumors showed decreased proliferation and increased apoptotic cell death (Supplementary Figs. S5 and S6).
SHP099/MEKi Combination Is Also Effective in TNBC and Serous Ovarian Cancer Models
Genetic (28, 29) and functional genomic (30, 31) analyses reveal striking similarities between TNBC and high-grade serous ovarian cancer (HGSC). These malignancies typically express WT RAS, and in some TNBC models MEK inhibition results in RTK upregulation and adaptive resistance (8). To explore the potential generality of combination MEK/SHP2 inhibition as a therapeutic strategy (and the utility of this combination in adaptive resistance to MEKi in WT RAS–expressing cells), we tested SHP099/MEKi combinations in TNBC and HGSC models.
Similar to its effects on KRAS-mutant cells, MEKi treatment increased RTK and RTK ligand gene expression in TNBC and HGSC lines (Supplementary Fig. S7A). SHP099 (10 μmol/L) alone had little effect on cell number or colony formation (Fig. 4A and B; Supplementary Fig. S7B). The MEKi AZD6244 or UO126 had variable single-agent effects, often (but not always) causing reduced cell proliferation compared with controls. Nevertheless, resistant cell populations were seen in almost all cell lines. The SHP099/MEKi combination showed increased efficacy, with additive to synergistic effects (Fig. 4A; Supplementary Fig. S7B; Supplementary Table S1). As in KRAS-mutant cell models (above), combination treatment slowed cell-cycle progression and enhanced cell death (Supplementary Fig. S7C and S7D). After 48 hours of single-agent treatment, SHP099 had little or no effect on RAS activation in any of the models (Fig. 4D). After MEKi treatment, however, RAS was hyperactivated to varying degrees in MDA-MB-468, HCC1954, CAL-120, and OVCAR-8 cells. In SHP099/MEKi–treated cells, RAS-GTP decreased to normal levels in randomly growing cells. These findings indicate that RAS activation is largely SHP2-independent under normal serum growth conditions, but is essential for the increase in RAS-GTP caused by the adaptive (RTK-driven) program evoked by MEKi treatment. As expected, 48-hour AZD6244 treatment led to increased MEK1/2 and ERK phosphorylation; consistent with its effects on RAS, SHP099 suppressed this increase (Fig. 4E), as well as ERK-dependent gene expression (Fig. 4F).
Finally, we treated mice bearing mammary fat pad xenografts derived from MDA-MB-468 or an extremely aggressive HGSC PDX with SHP099, trametinib, or both drugs. Single agents did not produce consistent regressions of MDA-MB-468 xenografts, and the ovarian PDX was highly resistant to both drugs. However, SHP099/trametinib caused substantial regression of MDA-MB-468 xenografts and markedly inhibited the growth of the HGSC PDX (Fig. 4G and H; Supplementary Fig. S4F), while significantly reducing tumor angiogenesis and cellularity in both models (Fig. 4I). Decreased tumor cell proliferation and increased apoptosis were observed in combination-treated tumors (Supplementary Fig. S6).
Discussion
Tumors evade targeted cancer therapies via an extensive repertoire of resistance mechanisms. One common theme involves activation of RTKs by inducing their expression and/or the expression of their ligands, which reactivates the inhibited pathway (5–9, 24, 32). Indeed, multiple, distinct sets of RTKs/RTK ligands were activated in response to MEKi treatment in the models that we tested. The heterogeneity of this adaptive response renders unfeasible combination therapies with MEKi and RTK inhibitors. However, targeting a common downstream component of RTK signaling in combination with MEKi might yield substantial efficacy. SHP2 has long been known to signal downstream of normal RTKs (10, 11), and cancer cells dependent on RTK activity are susceptible to SHP099 monotherapy (12, 13). We find that combined MEK/SHP2 inhibition blocks cell proliferation and promotes shrinkage of tumors with increased RAS/ERK pathway activation, including those that typically have WT RAS (TNBC and HGSC), as well as those driven by mutant KRAS (PDAC and NSCLC). Unexpectedly, our studies also shed new light on the long-elusive effect of SHP2 on RAS.
Although SHP099 reportedly has off-target effects in some cells (33), it is clearly “on target” in our experiments. We observed its expected biochemical effects on RAS/ERK pathway activation in the multiple lines tested. Moreover, two different drug-resistant mutants (PTPN11P491Q and PTPN11T253M/Q257L) rescue the effects of SHP099 in PDAC and NSCLC cells, respectively, whereas PTPN11 shRNA expression has similar biological and biochemical effects to SHP099. The MEKi used here also are highly validated, giving us confidence that the effects we observed reflect dual SHP2/MEK inhibition.
We expected SHP099 to block the adaptive increase in normal RAS activation that accompanies increased RTK signaling in MEKi–treated cells. Surprisingly, SHP099 also decreased mutant KRAS activation in MIAPaCA-2 cells, which only express KRASG12C, yet show clearly decreased KRAS activation following SHP099 treatment. KRASG12C has significant intrinsic GTPase activity (18), as exemplified by covalent RAS inhibitors that target KRASG12C-GDP (34). Hence, even though KRASG12C is largely refractory to RAS-GAPs, significant conversion to RAS-GDP must occur in cells via this intrinsic GTPase activity, and ongoing GDP/GTP exchange is required to maintain steady-state levels of KRASG12C-GTP. Other RAS mutants (except Q61 alleles) also retain some intrinsic GTPase activity, although less than does KRASG12C. SHP099 also led to decreased pERK levels in RAS-less MEFs expressing KRASG12D and KRASG12V in a manner linearly related to residual KRAS-GTPase activity. Recently, Nichols and colleagues (35) also reported variable effects of a new allosteric SHP2 inhibitor on mutant KRAS, although its chemical matter was not reported and its specificity was not established using drug-resistant mutants.
Genetic and biochemical analyses have firmly established that SHP2 acts upstream of RAS (10, 11), but whether it promotes exchange, inhibits GAP activity, or both has been controversial. Early work showed that SHP2, via its C-terminal tyrosine phosphorylation sites, can recruit GRB2/SOS (36, 37). A subsequent study reported diminished RAS exchange in lysates from cells expressing a GAB1 mutant that cannot bind SHP2 (38). However, multiple other reports claim that SHP2 antagonizes RAS-GAP by dephosphorylating its binding sites on RTKs or on SHP2-binding scaffolding adapters (39–41). Studies of Drosophila embryogenesis also argue for actions of the SHP2 ortholog, CSW, on the GAP binding sites in TORSO (42). Our findings, and those of Nichols and colleagues (35), show clearly that SHP2 acts upstream of SOS, and SHP2 inhibition can be bypassed by activated SOS, although we cannot exclude additional effects on RAS-GAP.
Single-agent SHP099 had little effect on 2-D proliferation or colony formation by cancer cells, but it significantly affected some xenografts and the KPC GEMM. There are several potential, non–mutually exclusive explanations for this apparent discrepancy. First, tumors occupy a hypoxic, nutrient-challenged, and potentially growth factor–deficient microenvironment; under such conditions, SHP2 might be essential for proliferation. Second, SHP2 might affect stromal support functions (e.g., growth factor production by cancer-associated fibroblasts, tumor angiogenesis). Tumor vascularity was decreased in all SHP099-treated xenografts, although whether this reflects direct inhibition of tumor angiogenesis or indirect effects on the tumor, with secondary effects on vessels, remains unclear. Third, SHP2 might affect the antitumor immune response, at least in the GEMMs. Interestingly, SHP099 and the drug combination had greater inhibitory effects in syngeneic mice than in nude mice (data not shown).
Our results comport with, and extend, previous studies of the effects of SHP2 modulation on other ERK pathway inhibitors. Prahallad and colleagues (14) found that SHP2 depletion (via PTPN11 shRNA or deletion) blocked adaptive resistance to the BRAFV600E inhibitor vemurafenib. They claimed that an SHP2 catalytic domain inhibitor (GS493) had similar effects, but that agent has off-target effects on tyrosine kinases (43). While this article was in revision, three independent groups reported that inhibition of SHP2 can sensitize KRAS-mutant or KRAS-amplified cancers to MEK inhibitors (44–46). Our results are in general agreement with their findings, although these reports used either the nonspecific SHP2 inhibitor GS493 (45), the earlier-generation MEKi AZD6244, which has a very short half-life in vivo and is not approved for cancer therapy (44), or mouse trametinib doses 4 times higher than the human MTD (45, 46). We show that SHP099 is “on-target” using drug-resistant SHP2 mutants and provide new evidence that combination therapy affects the tumor microenvironment (angiogenesis and stroma), can, at least in some models, promote differentiation of highly anaplastic tumor cells, and, when delivered for sufficient time, can prevent tumor regrowth after drug withdrawal. Taken together, all of these studies suggest that SHP2 inhibition might be a broadly applicable strategy to prevent or overcome adaptive resistance to kinase inhibition in a wide array of malignancies.
Methods
Cell Lines and Reagents
Cells were maintained in 5% CO2 at 37°C under the conditions described by the vendor or the source laboratory; details are available from C. Fedele or K.H. Tang upon request. Cells were tested at least every 3 months for Mycoplasma contamination by PCR (47) and genotyped by short tandem repeat analysis at IDEXX Bioresearch. See Supplementary Methods for details. SHP099 (HY-100388A) was purchased from MedChemExpress. Selumetinib-AZD6244 (S1008), UO126 (S1102), and trametinib (GSK1120212-S2673) were purchased from Selleckchem.
Plasmids, Retroviral and Lentiviral Production
Lentiviral and retroviral constructs were generated by standard methods (see Supplementary Methods). Viruses were produced by cotransfecting HEK293T cells with lentiviral or retroviral constructs and packaging vectors. Stable pools of infected cells were selected by using the appropriate antibiotic or by FACS for EGFP.
Cell Assays
Cell number was monitored by the PrestoBlue assay (Thermo Fisher). Potential drug synergy was determined by BLISS analysis as: Yab,P = Ya + Yb − YaYb, where Ya stands for the percentage inhibition of drug a and Yb stands for the percentage inhibition of drug b (48). For colony assays, cells (100–500) were seeded in 6-well plates and, after 16 hours, treated with DMSO or the indicated drugs. Colonies were stained with crystal violet, visualized by using the Odyssey Imaging System (LI-COR) and quantified with the ImageJ Colony Area PlugIn (49). For details, see Supplementary Methods. Cell-cycle distribution was monitored by flow cytometry using 7-AAD and analyzed by ModFit LT software (Verity Software House). Apoptosis was quantified by using the PE Annexin V Apoptosis Detection Kit (BD Biosciences).
Biochemical Assays
RAS activity was assessed by GST-RBD pulldown, followed by immunoblotting with pan-RAS or RAS isoform–specific antibodies. Whole-cell lysates were resolved by SDS-PAGE, followed by transfer to Nylon membranes. Immunoblots were performed with the indicated primary antibodies, followed by IRDye-conjugated secondary antibodies and visualization by LI-COR. For details, see Supplementary Methods.
IHC
pERK (Cell Signaling, 4370), CD31 (Cell Signaling, D8V9E), cleaved caspase-3 (Cell Signaling, D3E9), and Ki67 (Spring Biosciences, SP6) staining was performed on paraffin sections. OCT frozen sections were used for SA-βgal staining. Hematoxylin and eosin, Masson Trichrome, and PAS/Alcian Blue staining were performed by the Experimental Pathology Shared Resource at Perlmutter Cancer Center (PCC).
Animal Experiments
All animal experiments were approved by the NYU Langone Institutional Animal Care and Use Committee. Pancreas and lung cell line xenografts were established by subcutaneous injection of 5 × 106 cells in 50% Matrigel (Corning) into nude mice (nu/nu, #088 Charles River) MDA-MB-468 xenografts were established by injecting 5 × 106 cells in 50% Matrigel into the right lower mammary pad. Ovarian PDXs were established by injecting 5 × 105 cells in 50% Matrigel into the right lower mammary pad of NSG mice (The Jackson Laboratory). KPC 1203 cells (1 × 105 in Matrigel) were implanted into the pancreata of syngeneic male mice. Single agents and drug combinations were administered, and tumor size and body weight were monitored. For details, see Supplementary Methods.
qRT-PCR
Total RNA was isolated by the Qiagen RNeasy kit. cDNA was generated by using the SuperScript IV First Strand Synthesis System (Invitrogen). qRT-PCR was performed with Fast SYBR Green Master Mix (Applied Biosystems), following the manufacturer's protocol, in 384-well format in C1000 Touch Thermal Cycler (Bio-Rad). Differential gene-expression analysis was performed with CFX Manager (Bio-Rad) and normalized to GAPDH expression. Primers used are listed in Supplementary Table S2.
Statistical Analysis
Data are expressed as mean ± standard deviation. Statistical significance was determined using Student t test, Mann–Whitney U test, or one-way ANOVA. Statistical analyses were performed in Prism 7 (GraphPad Software). Significance was set at P = 0.05.
Disclosure of Potential Conflicts of Interest
B.G. Neel has ownership interest (including stock, patents, etc.) in Navire Pharma and is a consultant/advisory board member for the same. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions
Conception and design: C. Fedele, H. Ran, B.G. Neel, K.H. Tang
Development of methodology: C. Fedele, H. Ran, K.H. Tang
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): C. Fedele, B. Diskin, W. Wei, J. Jen, M.J. Geer, K. Araki, D.M. Simeone, K.H. Tang
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C. Fedele, H. Ran, J. Jen, M.J. Geer, U. Ozerdem, G. Miller, B.G. Neel, K.H. Tang
Writing, review, and/or revision of the manuscript: C. Fedele, U. Ozerdem, D.M. Simeone, B.G. Neel, K.H. Tang
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C. Fedele, H. Ran, K. Araki, K.H. Tang
Study supervision: C. Fedele, B.G. Neel, K.H. Tang
Other (review and interpretation of histopathology slides as a pathologist): U. Ozerdem
Acknowledgments
We thank Drs. Alec Kimmelman, Dafna Bar-Sagi, Douglas A. Levine, Gottfried E. Konecny, Robert Rottapel, and Kwok-Kin Wong for cell lines; Drs. Dafna Bar-Sagi, Jason Moffat, and David Root for plasmids; and the PCC Experimental Pathology and Precision Immunology shared resources for technical support. We also thank Dr. Toshiyuki Araki for advice and discussion on this project. This work was supported by R01CA49152 to B.G. Neel and R01CA131045 to D.M. Simeone. This work was supported by NIH Research Project Grant Program R01 CA49152 and CA131045.